Some cartilage tumors have epicenters within bone (enchondroma, chondromyxoid fibroma [CMF], chondrosarcoma), others have a predilection for the epiphysis (chondroblastoma), surface (juxtacortical chondroma, periosteal chondrosarcoma), or metaphyseal surface pointing away from the joint (osteochondroma), and still other types are complications of various underlying conditions. Usually, the age, site, and roentgenographic appearance indicate whether a cartilaginous lesion is benign or malignant, but at times the diagnosis is difficult and microscopic pathology critical. Histopathology alone may even have limitations in the prediction of malignant growth. Cartilaginous tumors are characterized by lobulation, bone endosteal scalloping, and punctuate, annular, or “rings,” “arcs,” and comma-like calcifications in the matrix. On MRI, the presence of lobules of cartilage 2 to 5 mm in diameter with or without calcification and/or ossification is relatively specific for well-differentiated cartilage matrix tumors. Chondroid tumors characteristically are bluish gray in color and are rich in extracellular matrix and stain positively with aldehyde-fuchsin at pH 1 and for S100 protein, a nonspecific mesenchymal marker, and SOX-9, a more specific marker of chondrocytic differentiation.
Normal chondroblasts and chondrocytes are identified histopathologically by having a rounded or multilobulated nucleus within a clear perinuclear space bathed in abundant extracellular matrix, often with an indistinct border. Cartilage is sparsely vascular. On electron microscopy, nuclear chromatin is evenly dispersed, with small and central nucleoli. A nuclear fibrous lamina is often prominent and characterized by abundant branching or dilated rough endoplasmic reticulum. There are few small lipid droplets but abundant glycogen and mitochondria, with a diffuse network of intermediate filaments. The cell membrane has microvilli and spikelike projections but no definitive external lamina (1).
Tumors in the hands and feet and on the surface of bone are usually benign. Synovial chondromatosis is almost always benign. Despite some tendency to malignancy, the overwhelming number of lesions in Ollier disease and Maffucci syndrome are benign. In general, several clinical, roentgenographic, and histopathologic findings may be helpful in distinguishing benign from malignant cartilage (2,3) (Table 10.1). No one factor is defining. Rather, a constellation of certain findings favors a particular interpretation. Beyond microscopy, flow cytometric analysis of the DNA content and patterns of cells has been applied in diagnostic studies of cartilage (see Appendix II). In one such study of 15 cartilage tumors, more than 90 percent of cells in lesions considered benign by conventional histologic analysis were diploid, and no cells were aneuploid. In the histologic low-grade chondrosarcoma, more than 10 percent were tetraploid and none aneuploid, and in the high-grade chondrosarcomas, fewer than 70 percent were diploid and fewer than 20 percent aneuploid (4). These findings suggest an adjunctive but not definitive role in the diagnosis of cartilage tumors by flow cytometry.
Investigators continue to search for tissue markers to help differentiate benign from malignant lesions. Tenascin, an extracellular matrix glycoprotein, is absent or found in small amounts in enchondroma and low-grade chondrosarcoma, but is increased in high-grade chondrosarcoma. Tenascin-C may stimulate invasive potential of chondrosarcoma cells via upregulation of MMP-1 expression (5). Chondrosarcomas contain cell nuclear antigen proliferating (Ki-67) at a higher rate and intensity (5). A prototype tumor-suppressor gene that exerts negative control of the cell cycle, p53, has been found to be more frequent in high-grade cartilaginous tumors and rarely positive in enchondromas. Elevation of p53 suggests a compensatory production to offset the tumor promoting effect of its nonmeasurable mutation (6).
TABLE 10.1 Cartilage Tumors
Benign
vs.
Malignant
Young persons
<
Adults (>50 years old)
Women
<
Men
Clinical
Pain related to other pathology such as fracture
Steady pain in absence of trauma
Small lesions
Large lesion >10 cm (>4 cm in fibula)
Hands and feet
Axial locations
Big bones
Imaging
Limited growth
Growing as ascertained by imaging studies
Diaphyseal
<
Metaphyseal and epiphyseal
Well-defined or sclerotic margins
Poorly defined margins
Irregular distribution of calcification
Confined within bone
Extension into soft tissue
Soft tissue mass
<1/2 of cortex is scalloped (if at all)
Excessive scalloping and thickening of the inner surface of the cortical bone
Confluent borderless masses of cartilage permeating marrow and intervening marrow demonstrating changes including myxoid and increased vasculature
Partial or complete encasement by trabecular lamellar bone conforming to cartilage outlines
Loss of encasement of cartilage by surrounding trabecular bone
<3/4 of cartilage encased in bone
No trapped lamellar bone
Cartilage masses trapping host trabecular bone and overriding the bone spicules
Bands of fibrosis between confluent cartilage lobules Cortical bone thickening
Infiltration of cortical Haversian bone and infiltration of soft tissue
Cellularity
<25 cells/400×
(Variable but) >100 cells/400×
Cytology
Occasional organized chondrocyte clusters
Increased and irregular distribution of cells throughout
Abundant binucleate cells > 26 per 20 high-power fields
Occasional binucleate cells
Multinucleated cells
Large bizarre hyperchromatic cells
Myxoid change
Cystic change
Mitoses
Occasional mitoses (<1/50 hpf)
Mitoses >2/50 hpf and occasional atypical mitosis
No abnormal mitoses
Homogeneous often small, dark nuclei
Pleomorphic nuclei and prominent nucleoli Nuclei irregular in size and shape
Cell ballooning
Vacuolization of cytoplasm
Special studies
+/- Ki-67 stain
++ Ki-67 stain
Elevated parathyroid hormone-related protein
Del Rosario et al. (7) reported that hyaline globules, spherical intracytoplasmic eosinophilic droplets, may be seen more frequently in low-grade chondrosarcomas.
Other investigators have analyzed chondroid tissue for genetic abnormalities. Although chromosomal rearrangements and translocations have been demonstrated, there is considerable cytogenetic heterogeneity, which limits diagnostic usefulness at this time (8,9).
Salient features of cartilage tumors are displayed in Figure 10.1.
Osteochondroma
Osteochondroma is a common type of exostosis (surface bone growth) characterized by the following features:
Exophytic (pedunculated or flat) growth on the surface of bone
A cartilaginous cap covered with a fibrous band
Contiguity with underlying cortical and trabecular bone and marrow
Cartilage in the cap undergoing endochondral ossification
The numerous types of osteochondromas that have been described include the following:
Bizarre parosteal osteochondromatous-like proliferation of bone (Nora lesion or BPOP)
Subungual exostosis
Para-articular (soft tissue) osteochondroma
Solitary Osteochondroma
Solitary osteochondromas are thought to arise in the peripheral zone of the growth plate when epiphyseal cartilage abnormally separates from the growth plate, setting up a satellite focus of endochondral ossification. An analogy with endometriosis, in which separate foci of endometrial tissue continue to respond to growth factors, can be made. Trauma to or deficiency of the perichondral ring has been suggested as a cause, which results in cartilage of the growth plate slipping to the surface of the bone and subsequently acting as an independent focus of growth. In one sense, osteochondromas represent a developmental abnormality rather than a true neoplasm. Once they reach a certain size, they, like the growth plate, complete endochondral ossification and, usually at the time of general skeletal maturation, cease growing. These lesions are quite common, but their true prevalence is difficult to assess because most are probably asymptomatic. A solitary lesion is six times more common than multiple hereditary exostosis (MHE).
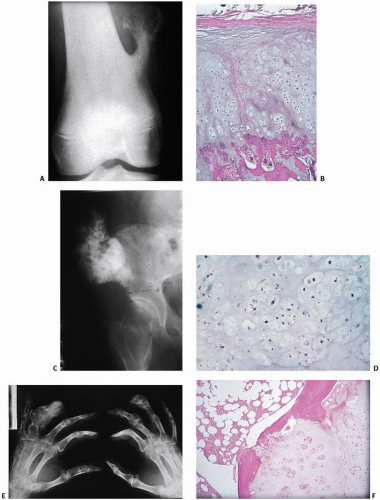
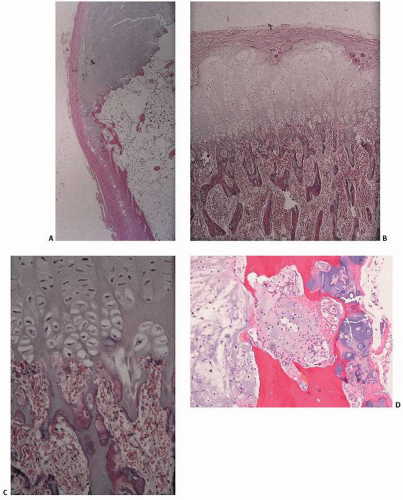
FIGURE 10.1. Cartilage lesions at a glance. (A, B) Osteochondroma. (A) Pedunculated metaphyseal mass pointing away from the knee joint. (B) Cartilage cap undergoing endochondral ossification. (C, D) Chondrosarcoma arising in multiple hereditary exostosis. (C) Irregular exophytic pelvic mass. (D) Chondroid lesion with disorganized cellularity compared with B. (E, F) Ollier disease. (E) Multiple expansile lytic lesions. (F) Proliferating cartilaginous nodules. (Continued)
FIGURE 10.1. (Continued) (M, N) Chondroblastoma. (M) Well-circumscribed lytic lesion extending to the end of the femur. (N) Hypercellular lesion with giant cells and waxy chondroid foci. (O, P) Chondromyxoid fibroma. (O) Trabeculated lytic lesion. (P) Mixture of chondroid, myxoid, and fibrous matrices. (Q, R) Chondrosarcoma. (Q) Calcified lesion with regions of lucency and erosion of the cortex. (R) Pleomorphic chondrocytes. (Continued)
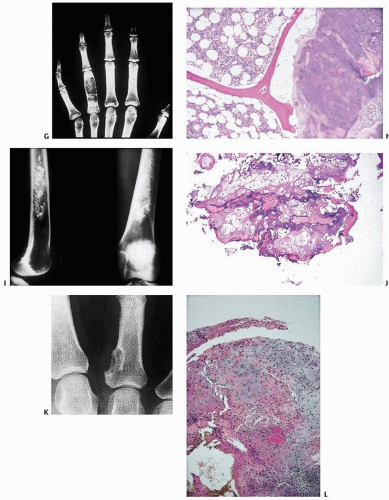
FIGURE 10.1. (Continued) (S, T) Dedifferentiated chondrosarcoma. (S) Calcified humeral lesion with extensive lytic destructive changes. (T) Colliding zones of chondrosarcoma and a malignant spindle cell tumor. (U, V) Clear cell chondrosarcoma. (U) Lytic lesion extending to the end of the proximal femur. (V) Chondroid matrix with vacuolated “clear” cells. (W, X) Mesenchymal chondrosarcoma. (W) Calcified lytic lesion in the mandible. (X) Primitive small cell population embedded in a waxy chondroid matrix.
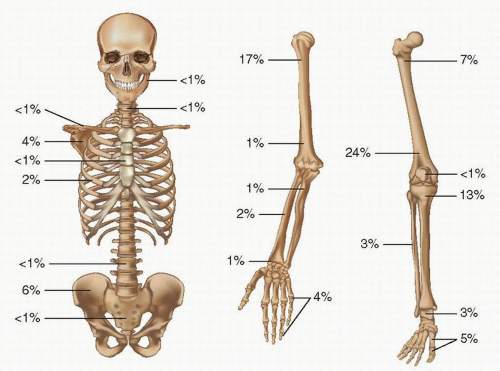
FIGURE 10.2. Sites and percentages of distribution in large reported series of osteochondroma.
Osteochondromas usually arise in the metaphysis of long bones; more than 50 percent of them arise in the distal femur (the most common site), proximal humerus, and proximal tibia (Fig. 10.2). If its distal and proximal metaphyses are included, the femur is by far the most common site. As might be expected in lesions thought to be caused by abnormal endochondral ossification, they are not found in the skull.
Clinically, osteochondromas are usually asymptomatic, but when they are detected, it is usually in male adolescents. The typical history is one of a long-standing mass, which may be painful. In roughly two-thirds of cases, diagnosis is made before the age of 20 years. Rarely, pedunculated osteochondromas on long stalks may fracture. They may cause adjacent bones to fracture, impinge on nerves, cause aneurysms, spontaneously infarct, separate and form osseocartilaginous loose bodies, or form bursae that may develop into osseocartilaginous loose bodies.
Surgery is usually indicated for (a) pain resulting from fracture of the lesion; (b) impairment of articular function and motion including locking of the knee; (c) excessive size; (d) unsightly deformity; (e) pressure on neurovascular structures (≈10 percent); (f) sudden increase in size, suggestive of malignant transformation; (g) development of a painful exostotic bursa; and (h) malignant transformation (2,10). Some advocate that, if surgery is indicated, removal of the osteochondroma after skeletal maturity will minimize the risk of recurrence.
Other problems with multiple osteochondromas include growth disturbance, such as angular deformities or uneven leg length; secondary impingement on nerves, vessels, or tendons (11); spinal cord compression; pleural irritation; and formation of bursae with pain and swelling (12,13).
Roentgenographically, these slow-growing lesions appear radiodense, pedunculated, and stalklike (exostosis), or broad-based, flattened, and sessile (Fig. 10.3). Osteochondromas are extensions of cortical and cancellous bone. They extend into the soft tissue, pointing away from the nearest joint, a feature thought to reflect the epiphyseal growth of bone away from the osteochondroma and/or the pull of adjacent tendons. The surface of the osteochondroma may be radiolucent or bullous with extensive calcification. Size is variable, from millimeters to more than 5 cm. Sessile or broad-based osteochondromas, if imaged by roentgenography en face, may deceptively suggest a circumscribed intraosseous lytic lesion. In these cases, the “lytic” appearance is caused by the x-ray beam passing through the cartilage (lytic cap) into the host cancellous bone.
Computed tomography (CT) will establish the cortical and medullary continuity between the osteochondroma and host bone, an important feature in the differential diagnosis of lesions such as periosteal or juxtacortical chondromas, which have an interface with cortical bone.
Magnetic resonance imaging (MRI) more clearly identifies and delineates the dimensions of the cartilage cap (T1, mixed signal; T2, high-intensity signal) (14). Bone scans in active osteochondromas are hot, reflecting the mineralization activity of endochondral ossification in the cap, a phenomenon that is helpful in excluding multiple lesions.
FIGURE 10.3. Radiographic appearance of osteochondroma. Lesions are most common in the metaphysis of long bones and may be pedunculated (A) or flat (B).
Ultrasound has been used to measure cap thickness (15).
Grossly, osteochondromas have a bosselated or smooth bluish-gray (cartilaginous) cap covered by a thin fibrous band (Fig. 10.4). The cap is usually 2 to 3 cm in thickness and has a smooth surface. It may be larger during adolescent growth. The cap usually ossifies after skeletal maturation. Whether osteochondromas are pedunculated and stalklike or broad-based, their cortical shells blend into the host cortical bone, and the base consists of marrow and cancellous bone confluent with the host marrow. Usually, no cortical bone is present at the base of an osteochondroma.
Microscopically, thin, periosteal, fibrous connective tissue covers a smooth or lobulated cartilaginous zone, which varies in thickness (Fig. 10.5). Columns of cartilage cells undergoing endochondral ossification are usually noted, although the transformation to bone may be rather abrupt. In the cartilage zone, the spatial distribution of chondrocytes may be irregular, and nuclear atypia may be present, but in nearly all cases, orderly cartilaginous columnar progression is discernible. Foci of necrosis may be seen.
The central core of an osteochondroma below the zone of endochondral ossification contains marrow (fatty and hematopoietic) and mature trabecular bone. Satellite zones of nonendochondrally ossified residual cartilage may be seen.
Sarcomas may arise in solitary osteochondromas, albeit rarely (<1 percent). They are usually chondrosarcomas, but other sarcomas, such as osteosarcomas, are well documented. Dedifferentiation of a chondrosarcoma can occur, but is extremely rare. In malignant osteochondromas, several features are suggestive of malignant change (Table 10.2).
The thickness of the cartilage cap has been used as an indicator of the likelihood of malignant transformation with a cutoff of 2 to 3 cm. Although cap thickness is easily measured when small and uniform, reliable measurements are more difficult when the cap is irregular or convoluted. In one study utilizing MRI measurement of the cartilage cap, the average cartilage cap thickness in malignant cases was 5 cm with a range of 2 to 12 cm (16).
The cap has typical MRI features of well-differentiated hyaline cartilage: low-intermediate S1 on T1W images, and markedly hyperintense on T2W images best for distinguishing the cap from adjacent muscle. Hypointense internal septa are commonly identified. The cap may appear very heterogeneous in the younger individual, while it is still undergoing active endochondral ossification (14).
Ultrasound has also been shown to be of value in assessing cap thickness. The cartilage cap appears hypoechoic with areas of punctuate or linear hyperechogenicity representing matrix calcification.
Radiation-Induced Osteochondroma
Lesions similar morphologically to osteochondromas occur in approximately 10 percent of patients undergoing radiation of bone in childhood. Lesions develop 1 year to many years after a dose of 1,600 to 6,425 rads (10,17). As with all bone irradiation, sarcomatous change is possible. Osteochondromas have been reported after total body irradiation (17). Children under 5 years are particularly at risk, as is exposure to approximately 1200 cGy. The effect of radiation in the formation of some osteochondromas is thought to be related to growth, with either arrested or disorganized and aberrant growth of endochondral foci in the growth plate the result.
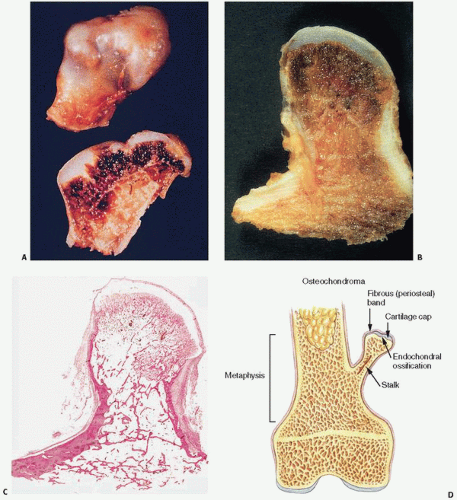
FIGURE 10.4. Gross appearance of osteochondroma. (A) Red marrow is directly beneath the bosselated, bluish gray cartilaginous cap, indicating hematopoiesis in the area of endochondral ossification. (B) A segment of the underlying host bone is directly contiguous with cortical bone and cancellous bone. (C) Microscopic appearance. (D) Osteochondroma. A pedunculated, cartilage-capped stalk located in the metaphysis pointing away from the joint space is the classic configuration.
FIGURE 10.5. Microscopic appearance of osteochondroma. Covered by periosteum (A), osteochondromas mimic endochondral ossification in the growth plate, with proliferating columns of cartilage undergoing conversion to calcified cartilage and ultimately bone (B, C). Chondrosarcoma arising in an osteochondroma showing increased atypical chondrocytes invading bone (D).
TABLE 10.2 Features Indicative of Malignant Transformation of Osteochondroma
Surgical
Difficult anatomic dissection of cap from surrounding soft tissue (adherence to soft tissue)
Clinical
Pain
Larger tumor (>6 cm)
Growth, especially rapid growth
Axial skeletal location
Roentgenographic
Variable pattern of mineralization
Radiolucent zones within the osteochondromatous cap
Soft tissue mass
Thick and irregular cartilage cap (>3 cm)
Indistinct margin
Cartilage foci beyond the periosteal cartilage cap
First described by Berger in 1814, this autosomally dominant genetic disorder is characterized by the formation of multiple osteochondromas. The overall prevalence has been estimated at 1 in 50,000, with approximately 10 percent of patients showing a family history (18). Approximately 50 percent of descendants are affected by 3.5 years of age, and nearly 100 percent by age 12 (18,19,20). Disturbances in the rate of endochondral ossification near affected sites cause uneven length of limbs; deformities of the forearm (>80 percent of patients), wrist, knee, and ankle (wrist and ulnar deviation, ankle valgus, genu valgum); shortening and bowing of the ulna and fibula; and tilt and tapering of the distal radial and tibial apophyses and distal radioulnar and tibiofibular diastasis (11) (Fig. 10.6). Shortened phalanges, broadened bones with flared metaphyses, ulnar shortening, radial head dislocation, carpal slips, “licked candy sticking” between radius and ulna, and scoliosis may also be seen (12). The condition is diagnosed after the age of 2 years when palpable masses are noted. More than 80 percent of cases are discovered in the first decade of life. The median age at diagnosis in a large pedigree study has been estimated at 3 years (range, birth to 12 years) (18).
Osteochondromas may affect several sites in one bone or multiple sites in multiple bones (21) (Fig. 10.7). Bilateral symmetric involvement is frequently encountered. Whereas the ilium is far less frequently affected than the femur by solitary osteochondromas, the ilium is frequently involved in MHE.
Malignant transformation has been estimated to occur in 5 percent to 28 percent of cases of MHE seen at referral centers (Table 10.3). It is certainly far more common in MHE than in symptomatic solitary osteochondroma (estimated at below 1 percent), and it preferentially affects flat bones (Table 10.3; Fig. 10.8). Because most reports of malignancy arising in MHE are from referral centers, a true incidence is difficult to know (19,20). Some estimates of sarcomatous change would suggest a lifetime risk of at least 2 percent to 4 percent, which typically occurs in the third to fifth decades of life, suggesting an annual risk of the development of sarcoma in this age group to be 0.1 percent. This risk is likely doubled in EXT 1 mutation carriers, roughly 45 percent of MHE patients. Chondrosarcoma is the most common malignancy described, but other sarcomas and even dedifferentiated chondrosarcomas have been reported (21).
An excellent animal model for human disease is seen in the horse (22). Several other animal species are also affected. Theories attempting to explain the etiology of MHE are numerous (11). Normal osteogenesis may cause isolation of cartilage nests, which then fragment and evolve into independent zones of endochondral ossification and leave an incomplete growth plate, thus accounting for the diminished growth potential of the affected bone. Alternatively, in some form of embryologic malformation, cells with a bone-forming mesenchyme potential are aberrantly placed. In close anatomic association with the periosteum, an abnormal signal may be sent to cells of the metaphyseal periosteum. Perhaps a defect in the periosteal ring at the periphery of the growth plate allows a type of herniation of cartilage cells, which escape normal remodeling.
MHE is inherited as an autosomal dominant disorder with variable penetrance (23). It has been linked to a family of EXT genes, which if mutated can lose heterozygosity and potentially cause osteochondromas. Mutations of the following genes have been shown to be associated with MHE:
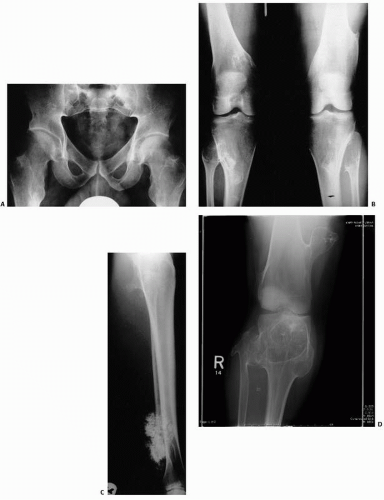
FIGURE 10.6. Multiple hereditary exostosis (roentgenographs). (A) In a frontal view of the pelvis, multiple flat exostoses in the femoral neck result in widening of the bone. (B) Frontal view of both knees reveals multiple osteochondromas arising from the metaphyses of the femora and tibiae. (C) An osteochondroma arising in the proximal tibia is in contradistinction to the radiodense, less circumscribed, proliferating low-grade chondrosarcoma arising in the lower tibia (D). MHE showing a loosened appearance in the femur, which is typically seen when a radiographic beam is directed into the cartilage cap.
a Incidence of sarcomatous transformation in multiple hereditary exostosis.
EXT 1 gene—a ubiquitously expressed gene on chromosome 8q23-q24, which encodes for a 746 amino acid protein related to tumor-suppressor function.
EXT 2 gene—a ubiquitously expressed gene located on chromosome 11p11-p13, which encodes a 718 amino acid protein linked to tumor-suppressor function.
EXT 3 gene—a gene located on chromosome 19p.
TRPS1 gene—a gene located on chromosome 8 near the EXT1 gene, which, if abnormal, may lead to cone-shaped epiphyses and, if deleted, the Langer-Giedion syndrome characterized by multiple exostoses, mental retardation, and craniofacial abnormalities.
EXT1 and EXT2 mutations account for roughly half and one-third of cases of MHE. The two genes, EXT1 and EXT2, encode ubiquitously expressed type-II transmembrane glycoproteins that are implicated in the elongation of the heparan sulfate-glycosaminoglycan (HS-GAG) chain of matrix proteoglycans (24). The role of HS-GAG in cell growth and differentiation signaling pathways reflects regulatory function of cartilage growth by EXT1/2 genes. The EXT1 gene is usually more often mutated than EXT2, with a variable prevalence among populations. The mutations cause premature termination of EXT proteins, inducing rapid inactivation and degradation with a nearly complete loss of their function. The effect of EXT1 mutations seems limited to growing bones. It has been suggested in some studies that patients with EXT1 mutations have a significantly worse condition than those with EXT2 mutations when analyzing stature, deformities, and functional impairment (24).
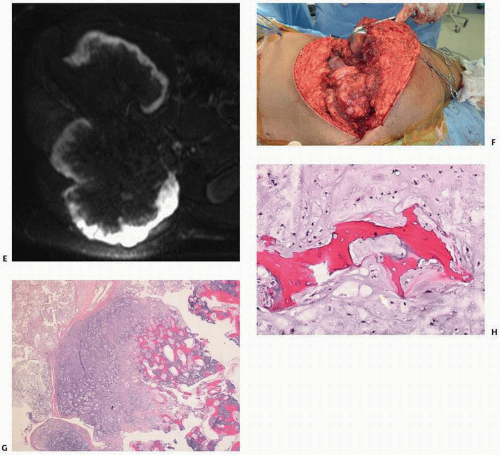
FIGURE 10.8. (A) Specimen radiograph of segmental resection of the ilium containing chondrosarcoma arising in an osteochondroma B through H: Radiograph of a19-year-old boy with a large solitary pelvic mass (B). CT (C) and reformatted images (D) showing a cauliflower-like growth. (Continued)
The Exostosis Genes and the Hedgehog Signaling Pathway
The EXT1 and EXT2 genes encode two glycosyltransferase subunits of the heparin sulfate (HS)-synthesizing system that elongates HS chains to specific proteins belonging to a class called proteoglycans. Mutations in the glycosyltransferase genes create truncated forms of the enzymes leading to lower enzyme activity and less HS chain synthesis. Chondrocytes isolated from persons with multiple hereditary exostosis contain less enzyme.
Cell surface HS and heparin sulfate proteoglycans (HSPGs) are required for signaling pathways such as fibroblast growth factor (FGF) and bone morphogenetic proteins (BMPs), and regulate the signaling range of IHH, signals that regulate the fine-tuning of chondrocyte proliferation and differentiation in the growth plate (25).
In long-bone growth plates, FGF signaling shortens cell column proliferation both by decreasing chondrocyte proliferation directly and by suppressing Ihh expression. BMPs needed for chondrocyte differentiation antagonize the effects of FGF signaling. Therefore, mutations in the EXT1 and EXT2 genes decrease HS synthesis, which reduces FGF signaling and leads to defects in chondrocyte differentiation.
FIGURE 10.8. (Continued) MRI shows a high focal signal intensity corresponding to the atypical cartilage cap (E). At operation, the nodularity of the cap is evident (F). Histology shows transformation of the cartilage cap (G) and foci of chondrosarcomatous encasement of bone (H).
Heparin sulfate proteoglycans can bind to hedgehog ligands, and, in the extracellular environment, this binding can control the region in which the hedgehog operates.
The hedgehog signaling pathway is crucial in the regulation of chondrocyte fate in the growth plate. Prehypertrophic chondrocytes in the growth plate express the Hh ligand Indian hedgehog (Ihh), which serves as a key regulator of endochondral ossification. Thus EXT mutations cause abnormal heparin sulfate proteoglycan processing, and this results in an abnormal diffusion of hedgehog ligand in the extracellular environment. The protein is not as able to effectively bind and control the movement of the hedgehog protein. Since hedgehog ligand patterns the terminal differentiation of growth-plate chondrocytes, the consequence of the mutation causes growth plate chondrocytes to pattern abnormally, resulting in the bone and cartilage growth occurring at an angle to the normal growth-plate direction. Alternatively, abnormal HSP may change the deformation and differentiation potential of perichondrial cells, resulting in the development of growth-plate-like cells that differentiate in an abnormal direction (25).
Thus exostoses develop juxtaposed to the growth plate and not within the growth plate.
MHE is a chronic disease causing a profound impact on the quality of life (26). Although pain is the greatest problem, the disorder has an extensive influence on daily activities, as well as on social and psychological well-being, causing significant disability. Among children, difficulty in continuing sports activities, difficulty in writing and using computers, and being bullied are of import. In adults, many need to change jobs or tailor their jobs to accommodate their disability.
Dysplasia Epiphysealis Hemimelica (Osteochondroma of the Epiphysis, Trevor Disease, Tarsoepiphyseal Aclasis)
Dysplasia epiphysealis hemimelica is a skeletal developmental disorder, usually manifested in early childhood, in which there is unilateral irregular enlargement of a medial epiphysis (27) (Fig. 10.9). Its incidence is approximately 1 in 1 million. The disorder most commonly involves the lower femur or upper tibia, distal tibia, and talus. Epiphyseal regions are most affected. Multiple epiphyses may be involved in two-thirds of the cases. The male-to-female ratio is 3:1.
Clinically, pain, uneven leg length, restricted motion, and deformities may be noted. Trevor disease is thought to be the result of fusion in the growth plate of isolated irregular centers of ossification, leading to asymmetric enlargement of the epiphysis (28). The resultant mass resembles an osteochondroma.
Pathologically, the excised lesion is reminiscent of an osteochondroma, with a cartilage cap and underlying zone of endochondral ossification and normal progression of cancellous bone formation.
Although benign in nature, varus or valgus deformities of the limb may ensue. When appropriate, surgical excision is the treatment of choice. Premature osteoarthritis may develop in the local joint.
FIGURE 10.9. Trevor disease (dysplasia epiphysealis hemimelica, tarsal aclasis). Radiographs of the elbow (A) and wrist (B) of the same patient demonstrate bony prominences in the olecranon process of the ulna and in the proximal carpal row, most likely in the triquetrum.
Although Trevor disease is generally considered a nongenetic transmitted disease, recent reports of an autosomally dominant transmitted disorder, termed hereditary epiphyseal osteochondromatosis, has been documented (29).
First described by Nora in 1983, bizarre parosteal osteochondromatous-like proliferations (BPOP) of bone are thought to be a distinct entity with similarities to a classic osteochondroma, except that the medullary portion of the osteochondroma is not necessarily directly in continuity with the medullary component of the host bone (30). BPOP arises from the surface of bone or the periosteum. It characteristically involves the small bones of the hands and feet or long bones during the third and fourth decades (Fig. 10.10). It is clinically significant in that it needs to be distinguished from malignant tumors of surface bone. If inadequately excised, the recurrence rate is high, approximately 55 percent in one series.
Histologically, BPOP is characterized by disorganized and hypercellular bone and cartilage, the cartilage often having a dark blue tinctorial quality (Fig. 10.10). Immunohistochemical studies of basic fibroblast growth factor (expressed in nearly all cells in the cartilage cap zone), vascular endothelial growth factor (expressed only in enlarged chondrocytes near osteocartilaginous interfaces), and chondromodulin-I transcripts (in the cartilage cap) have indicated similarities with endochondral ossification in a growth plate suggesting the lesion is a reparative process.
Similar lesions in different anatomic locations have been described, including subungual exostosis of the toe (vide infra) and florid reactive periostitis (Chapter 7).
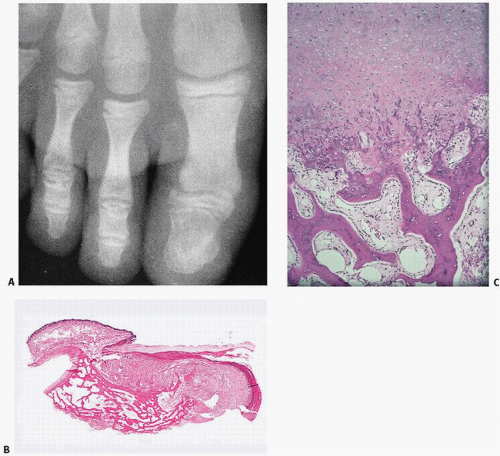
FIGURE 10.10. Nora lesion, BPOP (A) Roentgenographic features: a well-marginated mass of bone arises from the cortical surface of the affected phalanx of the finger. There is little alteration of the underlying bone. (B) Histopathology: fanlike proliferative cartilage (blue) blends into calcified cartilage and bone (pink) in varying degrees of organization. (C) Foci of endochondral ossification often reveal organized columns of cartilage.
A t(1;17)(q32;q21) has been reported as a unique recurring translocation identified in several cases. In addition, inversion of chromosome 7[inv(7)(q22q32) has also recently been described (31).
Subungual Exostosis
Subungual exostoses are common bony lesions of the foot, usually found under the nail on the distal dorsomedial aspect of the great toe (32). The distal phalanx of a finger may also be affected. There is often a convincing history of repetitive trauma or infection, suggesting a multifactorial etiology. Rarely larger than 1 cm in size, they may be painful or swollen and can ulcerate and become infected.
Roentgenographically, a hard bony mass is seen on the surface of the toe (Fig. 10.11). The lesion may be well defined, but is more apt to have ill-defined, hazy margins. It may or may not appear attached to the underlying bone.
Microscopically, the lesion has features of both fibrocartilaginous callus and an osteochondroma.
Usually, zones can be defined with adequate sectioning (Fig. 10.11). Superficially, proliferating fibrocartilaginous tissue blends with the adjacent soft tissue and may have areas resembling mineralizing callus. Closer to the toe, there is often a defined cartilaginous cap with zones of endochondral ossification maturing into bone, identical to what is seen in a classic osteochondroma. However, although the lesion may blend into the anatomic structure of the toe, the host bone cortex is usually intact. Proliferating peripherally, zones of this lesion may be quite cellular and overdiagnosed.
Surgical removal is curative, although the lesion may recur. There is no malignant potential.
The consistent finding in one study of a translocation (x;6) has suggested a neoplastic rather than reparative process.
Intra-Articular, Para-Articular, and Soft Tissue Osteochondromas
Osteochondroma-like lesions can occur in an intra-articular, para-articular, and soft tissue location (33). This is not surprising since synovial tissue, bursal tissue, and tenosynovial lining tissues are lined by cells that can undergo metaplasia to cartilage, which can subsequently form bone through endochondral ossification (34). These lesions are typically not connected to the underlying bone, a feature distinct from the classic osteochondroma.
FIGURE 10.11. (A) Roentgenograph of big toe with radiodense surface lesion in a patient with subungual exostosis. (B) Under low-power microscopy, the phalanx is seen in its entirety. Note nail (left) and bony growths on the surface of the phalanx connecting soft tissue and phalanx. (C) A fibrocartilaginous “cap” connects to the soft tissue and bone undergoing endochondral ossification in direct contact with the underlying phalanx.
Para-articular osteochondromas are rare osteocartilaginous tumors of soft tissue that behave in a benign fashion, but like synovial chondromas, they may mimic malignancy. Marginal excision appears adequate (33).
Posttraumatic Osteochondromas
The occurrence of a so-called posttraumatic intramedullary osteochondroma that has the histologic features of an osteochondroma with the exception of an inward growth into bone reflects the liberal use of the term “osteochondroma” in the literature (35).
Processus Supracondylaris Humeri
Processus supracondylaris humeri is a supracondylar process of the humus, an outgrowth of bone originating from the anterior-medial cortex of the distal humerus. It occurs in approximately 0.4 percent to 2 percent of whites and less frequently in blacks, and can measure up to 20 mm in length. It is a congenital human anatomical variation that may be related to a homologous structure normally found in amphibians, reptiles, and mammals.
Enchondroma
Enchondroma is the most common benign neoplasm of cartilaginous origin, the second most common if one considers the osteochondroma a neoplasm rather than a developmental disorder.
It is highly likely, since most enchondromas are asymptomatic, that they are far more common than diagnosed. Knee MRIs for other pathologies have documented nodules of cartilage (presumptively “incidental” enchondromas) in 2.9 percent of MRIs, most frequently in the distal femur (2 percent) (36).
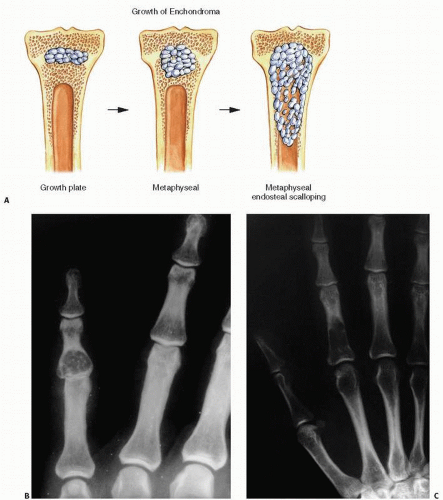
Classically, solitary enchondromas arise within the medullary cavity of bone, with more than half identified in the phalanges (approximately 53 percent found on the hand) (Fig. 10.12). Migration of enchondromas within long bones has been postulated on the basis of serial roentgenographic studies. Because they move from metaphysis to diaphysis (Fig. 10.13), enchondromas are thought to arise from the growth plate, much like osteochondromas, their surface, nontumorous, developmental counterpart.
Enchondromas most likely develop as a result of unregulated Hh signaling, whereby the normal downregulation of Hh signaling by Pth1h stimulation found in growth plate chondrocytes is disrupted (37).
FIGURE 10.12. Skeletal distribution of enchondromas.
Enchondromas may be clinically asymptomatic, but if not, they usually present early in life, between the ages of 10 and 30 years. There may be pain or swelling and even pathologic fracture in a weight-bearing bone.
Roentgenographically, enchondromas are well-defined, sharply demarcated, round or oval lesions with lobulation and thin and sclerotic borders (Fig. 10.14) (38). Usually located centrally within the metaphyseal portion of the bone, they may extend beyond this area. Mineralization in these lesions is variable, and calcification (and thus radiodensity) within them has been variably described as punctate, flocculent, and ringlike. Radiodense calcification, when present, may belie the real extent of tumor. Rare changes include soft tissue extension and periosteal reaction.
MRI reveals a well-marginated lesion that has low signal on T1-weighted sequences and high signal on T2-weighted sequences. Minimal marrow edema is seen about the periphery of the lesion, suggesting an indolent lesion.
Whereas management of conventional enchondromas is generally confined to observation, epiphyseal lesions are often symptomatic, and their proximity to the joint and propensity for pain and physeal involvement may require earlier definitive surgery (39) or biopsy to rule out chondroblastoma.
In short tubular bones, enchondromas are usually seen as well-defined lucencies, often located close to the metaphysis. The tufts of the distal phalanges are bones formed by membranous ossification, and, therefore, enchondromas should not be found at these sites. These lucencies may have areas of calcification. Calcification in an enchondroma is most common when the lesion occurs in a long bone or flat bone, such as a rib. The radiographic appearance of an enchondroma of long bone is almost similar to that of a bone infarction. However, the calcifications in enchondromas tend to appear close to the center of the lesions, whereas they are most often seen in the periphery of bone infarctions. Enchondromas are usually hot on a bone scan because of mineralization activity, the result of calcification of the cartilage rather than formation of bone.
Endosteal scalloping of the inner surface of the cortical bone is a common finding in enchondromas and is present in most cases in the hand (40). Cortical bone thickening is a more worrisome feature.
In long bones, enchondromas may be complicated, albeit rarely, by a secondary aneurysmal bone cyst (41).
Diagnosis of enchondroma is helped considerably by roentgenographic analysis. If the pathologist relies solely on histopathology, diagnosis may be difficult. However, there are both high-power microscopic and low-power microscopic characteristics that may be useful in distinguishing a benign from a malignant intramedullary cartilaginous tumor (Tables 10.1 and 10.4). In general, enchondromas appear as lobulated islands of benign cartilage encased by calcifying tissue and bone (Fig. 10.15). Examination of the bordering rim may reveal the mineralization progression of enchondral ossification or cartilage directly abutting on bone. Cellularity varies and may be associated with some irregularity, but, in general, nuclei are regular and uniform in distribution, with only focal myxoid or necrotic change. Although nucleoli and binucleated cells may be seen, they are not a prominent feature. Mitoses are rare. Cytoplasm is usually abundant. A lobular pattern of growth is maintained.
FIGURE 10.13. (A) Growth patterns of enchondromas. Most likely arising from growth plate cartilage, enchondromas grow circumferentially or in an ellipsoid tract down the shaft of the bone. (B) Roentgenographic changes show a centrally located, well-circumscribed lucency in the phalanx of the hand, which may or may not show calcifications. Large flocculent, calcified masses with rings and arcs are typically enchondromas in large bones such as the femur (C).
Benign-appearing asymptomatic enchondromas that are not weakening the bone can be watched. Curettage and bone grafting have been used in symptomatic cases, but if there is a pathologic fracture, surgery is usually delayed until healing is complete.
Variants of the classic intramedullary enchondroma are ones that are more eccentrically located in the bone (“eccentric enchondroma”), ones with a lobulated exuberant growth on the surface of the bone (“enchondroma protuberans”), and surface (periosteal or juxtacortical) chondromas. Cases of combined juxtacortical and intramedullary enchondromas are well documented.
Malignant change of an enchondroma is exceedingly rare, but well-documented cases have been reported (42).
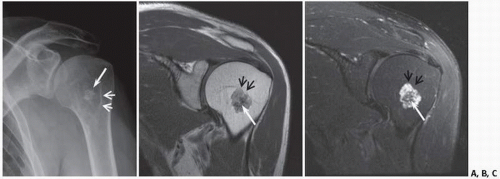
FIGURE 10.14. Low-grade chondral tumor. Frontal radiograph (x-ray) (A), oblique coronal proton density (B), and fat-suppressed MRI (C) show the typical appearance of an enchondroma (benign chondral tumor) with calcification (long white arrows). The noncalcified tumor outline (small arrows) is much more conspicuous on the MRI.
TABLE 10.4 Signs of Malignant Transformation of an Enchondroma (a Rare Event)
Pain in the absence of trauma
Older age of patient
x-ray film: extension into soft tissue
Metaphyseal enchondromas in long bones have been described in association with osteochondromas (43).
Metachondromatosis (MC) is a rare, heritable condition comprising multiple osteochondromas and multiple enchondromas first described by Maroteaux in 1971. The condition is inherited in an autosomal dominant pattern. Mutations in the PTPN11 gene have been found in some cases. Prevalence is unknown, and no ethnic predilection has been identified (43).
The location and orientation of the osteochondromas, as well as the lack of bone shortening and short stature, have been the mainstay of differentiating MC from MHE. Osteochondromas of MC point toward the joint to which they are adjacent, whereas osteochondromas of MHE point away. They have a predilection for the small bones of the hands and feet, and do not produce the skeletal growth disturbances and short stature associated with MHE. The osteochondromas have a tendency to regress or disappear after the first or second decade of life. Enchondromas associated with MC are characterized by involvement of the iliac crests and the metaphyses of long bones, particularly the proximal femur. Intraosseous striations can be seen in the proximal femur and irregular masses of “popcorn-like” chondroid calcification.
This “metachondromatosis” may represent a similar pathologic event occurring within bone and on its surface—that is, aberrant growth plate cartilage growing autonomously on the bone surface (osteochondroma) and synchronously intraosseous growth plate cartilage developing within the medullary canal, neither maturing as expected. Such combinations can give rise to extraordinary exophytic cartilaginous growths, sharing properties of both enchondromas and osteochondromas (44).
Chondrosarcoma arising in MC has been reported (45).
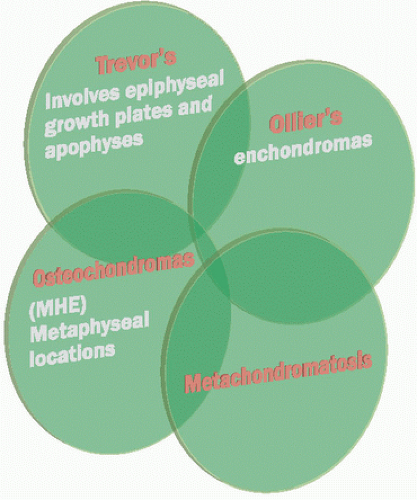
In fact, developmental disorders such as osteochondromas, Trevor disease, Ollier disease, and MC can overlap (46) (Fig. 10.16). The enchondromatous proliferation of an osteochondromatous growth has been termed enchondroma protuberans (47). Alternative views of this lesion are a periosteal chondroma (see Chapter 7), a low-grade chondrosarcoma, or an irritated, reactively proliferating cartilage cap of an osteochondroma. Nevertheless, some authors suggest an anatomically distinct lesion (48) (Fig. 10.17).
These distinctions are more than academic. Osteochondromas require only removal of the proliferating cap and stalk if benign. In enchondroma protuberans, cartilaginous foci within bone should be removed as well.
Ollier disease is a developmental abnormality in which multiple foci of cartilage, ranging from microscopic foci to bulky masses, may appear throughout the epiphyses, metaphyses, and diaphyses of the skeleton. Its incidence has been estimated at 1 in 100,000 (49). Clinically, patients with enchondromatosis show often asymmetric, unilateral involvement of the lower extremities, but any bone formed by endochondral ossification may be affected. The involved bones are usually shorter than normal, and there may be significant angular deformities and asymmetric involvement of metaphyses. The femur and tibia are the most commonly involved bones, but other bones, including the ilium and bones of the hands and feet, are also involved. Although the disorder is generally unilateral, it is often bilateral in the hands and feet. Ollier disease rarely affects bones formed by membranous ossification, such as the skull and facial bones.
Some of the roentgenographic characteristics of Ollier disease include abnormal patterns of radiolucency in distal and proximal ends of the long bones, often with asymmetric enlargement of the long bones and flaring at the metaphysis (Fig. 10.18). Enchondromas may calcify or even ossify, and cartilaginous proliferation has been described in all aspects of the skeleton. The younger the patient, the less likely it is for an enchondroma to be calcified. Although typically seen as cartilaginous islands in the metaphyses of long bones (Fig. 10.19), cartilaginous proliferation may be seen in medullary, cortical, periosteal, and even articular cartilaginous zones (Fig. 10.20). In fact, cartilaginous proliferation may occur on the surface, mimicking an osteochondroma.
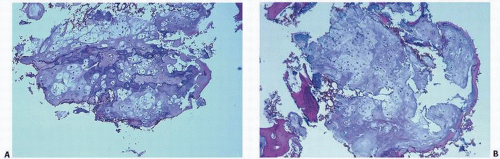
FIGURE 10.15. Histopathology of enchondroma. Bluish gray or slate gray lobules of proliferating chondrocytes with calcification in the periphery of the lobules (purple). The encasement of the lobules by calcification (purple in A) or bone (pink in B) is typical of a benign cartilaginous growth.
Histologically, Ollier disease is characterized by discrete or confluent lobules of proliferating, benign-appearing cartilage (Fig. 10.20). These lesions may or may not be calcified. In typical cases, unusual histologic features, such as mitotic activity, are not observed.
The exact cause of enchondromatosis is unknown. Although most cases of enchondromatosis are sporadic, families with multiple affected members have been reported, suggesting autosomal dominant inheritance with reduced penetrance. Alternatively, a random spontaneous mutation is possible, which might occur in early development, in mesoderm, therefore generating a mosaicism that will explain the propensity of multiple enchondromatosis to affect predominantly one side of the body in a similar way to what happens in GNAS mutation in the McCune/Albright syndrome.
Only gold members can continue reading. Log In or Register to continue