OBJECTIVES
After studying this chapter, youshould be able to:
Describe the neural mechanisms that control arterial blood pressure and heart rate, including the receptors, afferent and efferent pathways, central integrating pathways, and effector mechanisms involved.
Describe the direct effects of CO2 and hypoxia on the rostral ventrolateral medulla.
Define how the process of autoregulation contributes to control of vascular caliber.
Identify the paracrine factors and hormones that regulate vascular tone, their sources, and their mechanisms of action.
INTRODUCTION
In humans and other mammals, multiple cardiovascular regulatory mechanisms have evolved. These mechanisms increase the blood supply to active tissues and increase or decrease heat loss from the body by redistributing the blood. In the face of challenges such as hemorrhage, they maintain the blood flow to the heart and brain. When the challenge faced is severe, flow to these vital organs is maintained at the expense of the circulation to the rest of the body.
Circulatory adjustments are effected by altering the output of the pump (the heart), changing the diameter of the resistance vessels (primarily the arterioles), or altering the amount of blood pooled in the capacitance vessels (the veins). Regulation of cardiac output is discussed in Chapter 30. The caliber of the arterioles is adjusted in part by autoregulation (Table 32–1). It is also increased in active tissues by locally produced vasodilator metabolites, is affected by substances secreted by the endothelium, and is regulated systemically by circulating vasoactive substances and the nerves that innervate the arterioles. The caliber of the capacitance vessels is also affected by circulating vasoactive substances and by vasomotor nerves. The systemic regulatory mechanisms synergize with the local mechanisms and adjust vascular responses throughout the body.
| Vasoconstriction | Vasodilation |
|---|---|
| Local factors | |
Decreased local temperature Autoregulation | Increased CO2 and decreased O2 Increased K+, adenosine, lactate Decreased local pH Increased local temperature |
| Endothelial products | |
Endothelin-1 Locally released platelet serotonin Thromboxane A2 | Nitric oxide Kinins Prostacyclin |
| Circulating neurohumoral agents | |
Epinephrine (except in skeletal muscle and liver) Norepinephrine Arginine vasopressin Angiotensin II Endogenous digitalis-like substance Neuropeptide Y | Epinephrine in skeletal muscle and liver Calcitonin G-related protein Substance P Histamine Atrial natriuretic peptide Vasoactive intestinal polypeptide |
| Neural factors | |
| Increased discharge of sympathetic nerves | Decreased discharge of sympathetic nerves Activation of sympathetic cholinergic vasodilator nerves to vasculature of skeletal muscles of the limbs |
The terms vasoconstriction and vasodilation are generally used to refer to constriction and dilation of the resistance vessels. Changes in the caliber of the veins are referred to as venoconstriction or venodilation.
NEURAL CONTROL OF THE CARDIOVASCULAR SYSTEM
Most of the vasculature is an example of an autonomic effector organ that receives innervation from the sympathetic but not the parasympathetic division of the autonomic nervous system. Sympathetic noradrenergic fibers terminate on vascular smooth muscle in all parts of the body to mediate vasoconstriction. In some species, resistance vessels in skeletal muscles of the limbs are also innervated by vasodilator fibers, which, although they travel with the sympathetic nerves, are cholinergic (sympathetic cholinergic vasodilator system). These nerves are inactive at rest but can be activated during stress or exercise. Evidence for a sympathetic cholinergic vasodilator system in humans is lacking. It is more likely that vasodilation of skeletal muscle vasculature in response to activation of the sympathetic nervous system is due to the actions of epinephrine released from the adrenal medulla. Activation of β2-adrenoceptors on skeletal muscle blood vessels promotes vasodilation.
There are a few exceptions to the rule that only the sympathetic nervous system controls the vascular smooth muscle. The arteries in the erectile tissue of the reproductive organs, uterine and some facial blood vessels, and blood vessels in salivary glands, may also be controlled by parasympathetic nerves.
Although the arterioles and the other resistance vessels are most densely innervated, all blood vessels except capillaries and venules contain smooth muscle and receive motor nerve fibers from the sympathetic division of the autonomic nervous system. The fibers to the resistance vessels regulate tissue blood flow and arterial pressure. The fibers to the venous capacitance vessels vary the volume of blood “stored” in the veins. The innervation of most veins is sparse, but the splanchnic veins are well innervated. Venoconstriction is produced by stimuli that also activate the vasoconstrictor nerves to the arterioles. The resultant decrease in venous capacity increases venous return, shifting blood to the arterial side of the circulation.
When the sympathetic nerves are sectioned (sympathectomy), the blood vessels dilate. A change in the level of activity (increase or decrease) in sympathetic nerves is just one of the many factors that mediate vasoconstriction or vasodilation (Table 32–1).
The heart is one example of an effector organ that receives opposing influences from the sympathetic and parasympathetic divisions of the autonomic nervous system. Release of norepinephrine from postganglionic sympathetic nerves activates β1-adrenoceptors in the heart, notably on the sinoatrial (SA) node, atrioventricular (AV) node, His-Purkinje conductive tissue, and atrial and ventricular contractile tissue. In response to stimulation of sympathetic nerves, the heart rate (chronotropy), rate of transmission in the cardiac conductive tissue (dromotropy), and the force of ventricular contraction (inotropy) are increased. On the other hand, release of acetylcholine from postganglionic parasympathetic (vagus) nerves activates muscarinic receptors in the heart, notably on the SA and AV nodes and atrial muscle. In response to stimulation of the vagus nerve, the heart rate, the rate of transmission through the AV node, and atrial contractility are reduced.
The above description presents an oversimplified explanation of autonomic control of cardiac function. There are adrenergic and cholinergic receptors on autonomic nerve terminals that modulate transmitter release from nerve endings. For example, release of acetylcholine from vagal nerve terminals inhibits the release of norepinephrine from sympathetic nerve terminals, so this can enhance the effects of vagal nerve activation on the heart.
There is a moderate amount of tonic discharge in the cardiac sympathetic nerves at rest, but there is considerable tonic vagal discharge (vagal tone) in humans and other large animals. After the administration of muscarinic receptor antagonists such as atropine, the heart rate in humans increases from 70 beats/min, its normal resting value, to 150–180 beats/min because the sympathetic tone is unopposed. In humans in whom both noradrenergic and cholinergic systems are blocked, the heart rate is approximately 100 beats/min.
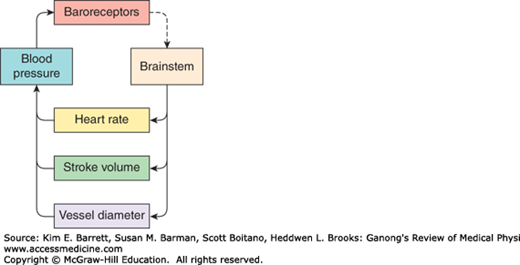
The cardiovascular system is under neural influences coming from several parts of the brainstem, forebrain, and insular cortex. The brainstem receives feedback from sensory receptors in the vasculature (eg, baroreceptors and chemoreceptors). A simplified model of the feedback control circuit is shown in Figure 32–1. An increase in neural output from the brainstem to sympathetic nerves leads to a decrease in blood vessel diameter (arteriolar vasoconstriction) and increases in stroke volume and heart rate, which contribute to a rise in blood pressure. This in turn causes an increase in baroreceptor activity, which signals the brainstem to reduce the neural output to sympathetic nerves.
FIGURE 32–1
Feedback control of blood pressure. Brainstem excitatory input to sympathetic nerves to the heart and vasculature increases heart rate and stroke volume and reduces vessel diameter. Together these increase blood pressure, which activates the baroreceptor reflex to reduce the activity in the brainstem.
Venoconstriction and a decrease in the stores of blood in the venous reservoirs usually accompany increases in arteriolar constriction, although changes in the capacitance vessels do not always parallel changes in the resistance vessels. In the presence of an increase in sympathetic nerve activity to the heart and vasculature, there is usually an associated decrease in the activity of vagal fibers to the heart. Conversely, a decrease in sympathetic activity causes vasodilation, a fall in blood pressure, and an increase in the storage of blood in the venous reservoirs. There is usually a concomitant decrease in heart rate, but this is mostly due to stimulation of the vagal innervation of the heart.
One of the major sources of excitatory input to sympathetic nerves controlling the vasculature is a group of neurons located near the pial surface of the medulla in the rostral ventrolateral medulla (RVLM; Figure 32–2). The axons of RVLM neurons course dorsally and medially and then descend in the lateral column of the spinal cord to the thoracolumbar intermediolateral cell column (IML). They contain phenylethanolamine-N-methyltransferase (PNMT; see Chapter 7), but glutamate is the excitatory transmitter they secrete to activate preganglionic sympathetic neurons. Neurovascular compression of theRVLM has been linked to some cases of essential hypertension in humans (Clinical Box 32–1).
FIGURE 32–2
Basic neural pathways involved in the control of blood pressure. The drawing shows a parasagittal section of the brainstem and its connections to the thoracic spinal cord. The rostral ventrolateral medulla (RVLM) is one of the major sources of excitatory input to sympathetic nerves controlling the vasculature. These neurons receive inhibitory input from the baroreceptors via an inhibitory (GABAergic) neuron in the caudal ventrolateral medulla (CVLM). The nucleus of the tractus solitarius (NTS) is the site of termination of baroreceptor afferent fibers that release glutamate. ACh, acetylcholine; GABA, γ-aminobutyric acid; Glu, glutamate; IML, intermediolateral cell column; NE, norepinephrine.

The activity of RVLM neurons is determined by many factors (see Table 32–2). They include not only the very important fibers from arterial baroreceptors but also fibers from other parts of the nervous system and from the carotid and aortic chemoreceptors. In addition, some stimuli act directly on RVLM neurons to increase their firing rate.
| Direct stimulation |
|---|
CO2 Hypoxia |
| Excitatory inputs |
Cortex via hypothalamus Mesencephalic periaqueductal gray Brainstem reticular formation Pain pathways Somatic afferents (somatosympathetic reflex) Carotid and aortic chemoreceptors |
| Inhibitory inputs |
Cortex via hypothalamus Caudal ventrolateral medulla Caudal medullary raphe nuclei Lung inflation afferents Carotid, aortic, and cardiopulmonary baroreceptors |
There are descending tracts to the vasomotor area from the cerebral cortex (particularly the limbic cortex) that relay in the hypothalamus. These fibers are responsible for the blood pressure rise and tachycardia produced by emotions such as stress, sexual excitement, and anger. The connections between the hypothalamus and the RVLM are reciprocal, with afferents from the brainstem closing the loop.
Inflation of the lungs causes vasodilation and a decrease in blood pressure. This response is mediated via vagal afferents from the lungs that inhibit RVLM and sympathetic nerve activities. Pain usually causes a rise in blood pressure via afferent impulses converging in the RVLM. However, prolonged severe pain may cause vasodilation and fainting. The activity in afferents from exercising muscles probably exerts a similar pressor effect via a pathway to the RVLM. The pressor response to stimulation of somatic afferent nerves is called the somatosympathetic reflex.
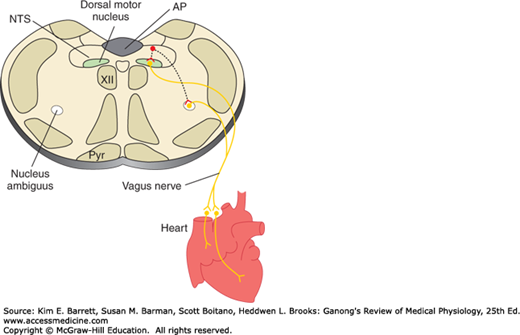
The medulla is also a major site of origin of excitatory input to cardiac vagal motor neurons in the nucleus ambiguus (Figure 32–3). Table 32–3 is a summary of factors that affect the heart rate. In general, stimuli that increase the heart rate also increase blood pressure, whereas those that decrease the heart rate lower blood pressure. However, there are exceptions, such as the production of hypotension and tachycardia by stimulation of atrial stretch receptors and the production of hypertension and bradycardia by increased intracranial pressure.
FIGURE 32–3
Basic pathways involved in the control of heart rate by the vagus nerves. Neurons in the nucleus of the tractus solitarius (NTS) project to and excite cardiac preganglionic parasympathetic neurons primarily in the nucleus ambiguus. Some are also located in the dorsal motor nucleus of the vagus; however, this nucleus primarily contains vagal motor neurons that project to the gastrointestinal tract. AP, area postrema; Pyr, pyramid; XII, hypoglossal nucleus.
| Heart rate accelerated by: |
Decreased activity of arterial baroreceptors Increased activity of atrial stretch receptors Inspiration Excitement Anger Most painful stimuli Hypoxia Exercise Thyroid hormones Fever |
| Heart rate slowed by: |
Increased activity of arterial baroreceptors Expiration Fear Grief Stimulation of pain fibers in trigeminal nerve Increased intracranial pressure |
The baroreceptors are stretch receptors in the walls of the heart and blood vessels. The carotid sinus and aortic arch receptors monitor the arterial circulation. Receptors are also located in the walls of the right and left atria at the entrance of the superior and inferior venae cavae and the pulmonary veins, as well as in the pulmonary circulation. These receptors in the low-pressure part of the circulation are referred to collectively as the cardiopulmonary receptors.
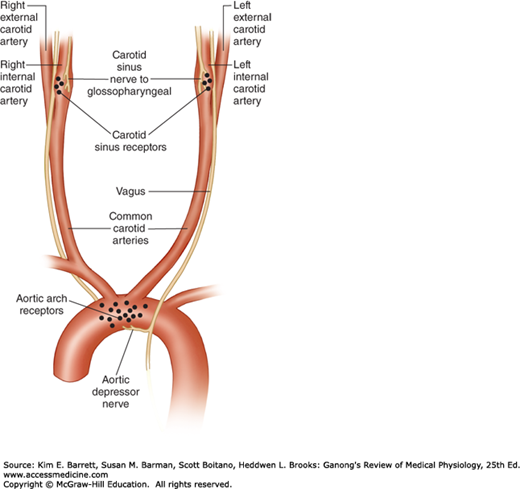
The carotid sinus is a small dilation of the internal carotid artery just above the bifurcation of the common carotid into external and internal carotid branches (Figure 32–4). Baroreceptors are located in this dilation. They are also found in the wall of the arch of the aorta. The receptors are located in the adventitia of the vessels. The afferent nerve fibers from the carotid sinus form a distinct branch of the glossopharyngeal nerve, the carotid sinus nerve. The fibers from the aortic arch form a branch of the vagus nerve, the aortic depressor nerve.
FIGURE 32–4
Baroreceptor areas in the carotid sinus and aortic arch. One set of baroreceptors (stretch receptors) is located in the carotid sinus, a small dilation of the internal carotid artery just above the bifurcation of the common carotid into external and internal carotid branches. These receptors are innervated by a branch of the glossopharyngeal nerve, the carotid sinus nerve. A second set of baroreceptors is located in the wall of the arch of the aorta. These receptors are innervated by a branch of the vagus nerve, the aortic depressor nerve.
The baroreceptors are stimulated by distension of the structures in which they are located, and so they discharge at an increased rate when the pressure in the carotid sinus and aortic arch rises. Their afferent fibers pass via the glossopharyngeal and vagus nerves to the medulla. Most of them end in the nucleus of the tractus solitarius (NTS), and the excitatory transmitter they secrete is glutamate (Figure 32–2). Excitatory (glutamate) projections extend from the NTS to the caudal ventrolateral medulla (CVLM), where they stimulate γ-aminobutyric acid (GABA)-secreting inhibitory neurons that project to the RVLM. Excitatory projections also extend from the NTS to the vagal motor neurons in the nucleus ambiguus and dorsal motor nucleus (Figure 32–3). Thus, increased baroreceptor discharge inhibits the tonic discharge of sympathetic nerves and excites the vagal innervation of the heart. These neural changes produce vasodilation, venodilation, hypotension, bradycardia, and a decrease in cardiac output.



