The first-line antihypertensive drug classes are thiazide diuretics (TZD), angiotensin-converting enzyme inhibitors (ACEIs), CCBs, and angiotensin receptor blockers (ARBs). β-Receptor blockers no longer enjoy a first-line status in the treatment of HTN, indicated by national guidelines released by the AHA in 2013. Among the remaining choices, a TZD should be included in most patients’ regimens because of low cost of TZDs and consistent evidence for their high efficacy across multiple controlled trials. Caution: TZD can precipitate gout attacks, and should be avoided in patients with known gout.
For most patients, each medication should be started at the lowest recommended dosage and titrated upward, if necessary, at 2- to 8-week intervals, depending on the severity of the HTN. Elderly patients may require even lower doses. Begin with a single-drug or a low-dose combination. More than one half of patients will require at least two medications to control their HTN. Dose titration and switching of medications are often both necessary. Patient education about this is important to engender trust and realistic expectations.
Follow-Up
Well patients with Stage 1 HTN should have follow-up visits every 1 to 2 months until the BP goal is reached without significant medication side effects (i.e., side effects that are unacceptable to the patient or the physician). Patients with Stage 2 HTN and/or complicating comorbidities should be seen every 2 to 4 weeks until the BP is clearly coming under control without unacceptable side effects. Once the BP goals are reached and stable on a given therapeutic regimen, follow-up can be stretched out to 6 months, unless other conditions dictate more frequent visits.
Goal BP levels should be SBP <140 mmHg and DBP <90 mmHg for most patients. However, in patient >59 years of age, the JNC-8 now has given the strongest of recommendations (Grade A evidence from randomized controlled trials), to treat to a goal of SBP of <151 and a DBP of <91. As mentioned previously, neither diabetics nor patients with CKD require any more aggressive BP goals than those of the uncomplicated hypertensive patient (140/90). Many clinicians choose to set target BP levels below these threshold because in population-based studies cardiovascular risk rises steadily with BP level, starting at SBP >115 mmHg or DBP >75 mmHg. However, care should be taken to avoid overly aggressive BP reduction in elderly patients, because they are at higher risk for orthostasis, tend to have stiffer arteries, and are at higher risk for stroke from episodic hypotension.
If treatment goals have not been met at the prescribed follow-up intervals, the medication dose should be changed, a different class of drug should be tried, or a second drug from another class should be added (see Table 9.1-1). Combining two first-line drugs from different classes at low to moderate doses is often effective, and including a diuretic is desirable. However, the exception to this is the use of a dual blockade method of the renin–angiotensin–aldosterone system. Several new randomized trials and a meta-analysis all showed that there was no benefit to the use of dual blockade (an ACE + an ARB), and indeed, may harm patients with or without heart failure. Avoid unwanted drug interactions, especially those that have cardiac and electrolyte effects. β-Blockers, central sympatholytics, α1-blockers, and peripheral antiadrenergics are best reserved as second-line drugs (except in pregnancy, as discussed below). Direct vasodilators are useful for patients failing treatment with first- and second-line drugs, but they should be combined with a diuretic.
Laboratory tests at follow-up are determined by the type of therapy, comorbid conditions, and the baseline values.
Special Therapy
Hypertensive crises are rare clinical emergencies in which high BP must be lowered immediately to prevent or limit a morbid complication. The situation, not the BP level alone, constitutes the emergency. Examples include:
• Acute pulmonary edema
• Acute MI
• Hypertensive encephalopathy
• Eclampsia
• Dissecting aortic aneurysm
In these situations, a controlled reduction of BP by 20% to 25% over a few minutes to a few hours is indicated.
Hypertensive urgencies are situations in which BP should be lowered to 160 to 170/100 to 110 mmHg within 24 hours to prevent complications. These include:
• Severe perioperative HTN
• Accelerated malignant HTN (BP >220/120 mmHg and rising)
Precipitous decreases in BP should be avoided. The goal is clinical stabilization, not normalization of BP. Relatively short-acting parenteral (IV) antihypertensives, followed by oral therapy, usually work best. Suggested IV drugs and doses are listed in Table 9.1-3. If IV therapy is not an option, oral captopril (Capoten), 25 mg, clonidine (Catapres), 0.1 to 0.2 mg, or labetolol (Normodyne), 200 to 400 mg, can be used; each has a hypotensive effect within 1 hour. Sublingual administration is not more effective than oral.
In the setting of acute cerebrovascular attack, HTN should generally not be treated unless SBP is greater than 220 mmHg or unless there are signs of progressive intracranial bleeding. Quiet bed rest often results in a significant decrease in BP.
Complications
Antihypertensive medications have variable effects on cardiac conduction, cardiac contractility, arterial and venous tone, renal function, and electrolyte metabolism (especially potassium). The prescriber must be aware of these potential side effects when deciding on therapy and during follow-up examinations. Drug interactions may potentiate or ameliorate symptomatic or metabolic side effects. New signs or symptoms of cardiac arrhythmia, dyspnea with exertion, edema, or fatigue should be thoroughly investigated. The serum electrolyte panel, BUN, and creatinine should be checked at least once per year; abnormalities should be addressed and followed up. Patients with existing cardiac disease, renal disease, or diabetes, and those on multiple medications, are at the highest risk for complications. Such patients also are likely to gain more absolute benefit from control of HTN than are patients without diabetes or TOD.
Parenteral Drugs for Hypertensive Crisis |
Drug name (trade name) | Dose for hypertensive crisisa |
Labetolol (Normodyne) | 20–40 mg IV q10 min |
Methyldopa (Aldomet) | 250–500 mg IV q6 h |
Hydralazine (Apresoline) | 20–40 mg IV q1–2 h nonpregnant 5–10 mg IV q20 min in pregnancy |
Diazoxide (Hyperstat) | 50–150 IV q15 min |
Enalaprilat (Vasotec IV) | 1.25 mg IV q6 h |
Nitroprusside (Nipride) | 0.2–10 µg/kg/min IV (use low dose in pregnancy) |
aReview of full prescribing information is strongly advised.
SPECIAL CONSIDERATIONS
Hypertension in Pregnancy
HTN occurs in 6% to 8% of pregnancies in the United States. It is associated with significant maternal morbidity, including seizure, stroke, encephalopathy, and hemorrhage. Additionally, HTN in pregnancy is a major contributor to uteroplacental insufficiency, placental abruption, prematurity, and fetal demise. Recently, the increasing prevalence of worldwide obesity and metabolic syndrome has led to concern for increasing numbers or women that could develop HTN in their childbearing years and pregnancy.
HTN in pregnancy is classified as either chronic or gestational. Criteria for diagnosis of either form are similar to the nonpregnant state:
• Mild chronic or gestational HTN is defined as SBP of 140 mmHg greater or DBP of 90 mmHg or greater
• Severe chronic HTN is defined as SBP of 180 mmHg or greater or DBP of 110 mmHg or greater
• Severe gestational HTN is defined as SBP of 160 mmHg or greater or DBP of 110 mmHg or greater
HTN in pregnancy should be diagnosed only after an elevated BP is documented on at least two readings taken 6 hours apart with the patient in the sitting position after a 10-minute rest. The HTN is considered chronic if the patient was diagnosed prior to conception or prior to the 20th week of gestation. Women with no recent BP readings who present for prenatal care after the 20th week, and who meet the criteria for HTN, should be considered to have gestational HTN. If HTN persists beyond the usual postpartum period, a diagnosis of chronic HTN can be made in retrospect.
Pre-eclampsia is a pregnancy-induced, multisystem disease defined by gestational HTN with proteinuria (2+ on dipstick on two occasions 6 hours apart or >3 g/24-hour urine collection). Given the increased risk of morbidity and mortality associated with pre-eclampsia, all women diagnosed with HTN during pregnancy should have a 24-hour quantitative urine protein measured. For those women with chronic HTN, a baseline measurement will document possible pre-existing renal disease that may influence subsequent diagnosis of pre-eclampsia. Chronic HTN is a known risk factor for the development of pre-eclampsia, with 20% to 30% of women with chronic HTN developing pre-eclampsia. The classification of pre-eclampsia as well as its treatment is covered in Chapter 14.8.
Treatment for chronic HTN in pregnancy and gestational HTN (without proteinuria) is dictated by the known effects of antihypertensive medications on uteroplacental blood flow and fetal outcome studies. Because numerous controlled trials have failed to demonstrate fetal or maternal benefit from treating mild HTN in pregnancy, the American College of Obstetrics and Gynecology (ACOG) recommends not starting antihypertensive medication for mild chronic or gestational HTN in pregnancy, unless there are comorbid conditions such as HTN-associated headaches, TOD, or rising BP levels.
For women already taking antihypertensive medication at the time of pregnancy diagnosis, current data support stopping therapy if HTN is mild, or switching treatment to the smallest effective dose of a first-line antihypertensive drug for use in pregnancy.
• First-line antihypertensive drugs for HTN during pregnancy
• Methyldopa 250 to 500 mg PO tid–qid
• Labetolol 100 to 400 mg bid–tid
• Acceptable second-line choices
• Other β-blockers (excluding atenolol that has been associated with growth restriction)
![]() Metoprolol 50 to 200 mg bid
Metoprolol 50 to 200 mg bid
![]() Pindolol 5 to 15 mg bid
Pindolol 5 to 15 mg bid
• CCBs
![]() Nifedipine 10 to 30 mg tid
Nifedipine 10 to 30 mg tid
![]() Nicardipine 20 to 40 mg tid
Nicardipine 20 to 40 mg tid
• Hydrochlorothiazide 25 to 50 mg qd
• Hydralazine 10 to 50 mg qid
• Contraindicated in pregnancy
• ACE inhibitors
• ARBs
• Aldosterone antagonists
The use of ACE inhibitors during pregnancy is associated with a variety of renal and pulmonary toxicities in the fetus.
Treatment of acute, severe HTN in pregnancy should occur expeditiously to reduce the risk of maternal stroke and placental abruption.
Hydralazine has been the preferred agent in the United States due to its long history of safety and rapid onset of action; however, thrombocytopenia has been rarely reported in neonates born to women treated in the third trimester.
• Recommended antihypertensive drugs for acute severe HTN in pregnancy
• Hydralazine 5 to 10 mg IV every 15 to 20 minutes
• Labetalol 20 mg IV bolus with 20 to 40 mg every 15 minutes as needed
• Nifedipine* 10 mg PO q15 minutes, max 30 mg
• Nicardipine* 5 mg per hour IV, increase at 2.5 mg per hour q5 to 15 minutes up to 15 mg per hour
• Sodium nitroprusside 0.25 μg/kg/minute IV, increase 0.25 μg/kg/min q5 minutes up to 5 μg/kg/minute
REFERENCES
1. James PA, Oparil S, Carter BL, et al. Evidence based guidelines for the management of high blood pressure in adults. Report from the panel members appointed to the Eighth Joint National Committee. JAMA 2014;311:503–520.
2. National High Blood Pressure Education Program. The seventh report of the Joint National Committee on prevention, detection, evaluation and treatment of high blood pressure (JNC 7). Bethesda, MD: NIH National Heart, Lung, and Blood Institute; 2003. Publication 03–5233.
3. ALLHAT Collaborative Research Group. Major outcomes in high-risk hypertensive patients randomized to angiotensin-converting enzyme inhibitor or calcium channel blocker vs diuretic: the Antihypertensive and Lipid-Lowering Treatment to Prevent Heart Attack Trial (ALLHAT). JAMA 2002;288(23):2981–2997.
4. Tronvik E, Stovner LJ, Hagen K, et al. High pulse pressure protects against headache: prospective and cross-sectional data (HUNT study). Neurology 2008;70(16):1329–1336.
5. Appel LJ, Champagne CM, Harsha DW, et al. Effects of comprehensive lifestyle modification on blood pressure control: main results of the PREMIER clinical trial. JAMA 2003;289(16):2083–2093.
6. New England Journal of Medicine. JW Gen. Med. December 31, 2013.
7. Dahlof B, Devereux RB, Kjeldsen SE, et al. Cardiovascular morbidity and mortality in the Losartan Intervention for Endpoint reduction in hypertension study (LIFE): a randomised trial against atenolol. Lancet 2002;359(9311):995–1003.
8. Demers C, McMurray JJ, Swedberg K, et al. Impact of candesartan on nonfatal myocardial infarction and cardiovascular death in patients with heart failure. JAMA 2005;294(14):1794–1798.
9. Casaa P, Weiliang C, Stavros L, et al. Effect of inhibitors of the renin-angiotensin system and other antihypersensitive drugs on renal outcomes: systematic review and meta-analysis. Lancet 2005;366;2026–2033.
10. Report on National High Blood Pressure Education Program Working Group on High Blood Pressure in Pregnancy. Am J Obstet Gynecol 2000;183:S1–S22.
11. Sibai BM. Diagnosis and management of gestational hypertension and preeclampsia. Obstet Gynecol 2003;102:181–192.
12. Lowe SA, Brown, MA, Dekken GA, et al. Guideline for the management of hypertension disorders in pregnancy 2008. J Obstet Gynecol 2009;49(3):242–246.
13. Yoder SR, Thornberry MD, Bisognano JD. Hypertension in pregnancy and women of childbearing age. Am Med J 2009;122:890–895.
14. Chronic hypertension in pregnancy. ACOG Practice Bulletin No. 29. July 2001.
15. American College of Obstetricians and Gynecologists. Task Force on Hypertension in Preganancy. Hypertension in pregnancy. Report of the American College of Obstetricians and Gynecologists’ task force on hypertension in pregnancy. Obstet Gynecol 2003:122(5):1122–1131.
|
GENERAL PRINCIPLES
• Ischemic heart disease (IHD) is a chronic medical condition prone to acute exacerbations and affecting a sizable percentage of the adult population. The manifestations may be broad and have impact on inpatient and outpatient care. This chapter focuses on outpatient diagnosis and management.
• IHD is a term interchangeable with coronary artery disease (CAD), coronary heart disease (CHD), and atherosclerotic heart disease (ASHD). Although in the most precise sense the terms are not identical, generally they all refer to a condition of obstructed blood flow in the coronary arteries that may result in ischemia or infarction of the myocardium. Clinically, patients with IHD may be asymptomatic, may have chronic stable angina, may present with an acute coronary syndrome (ACS; see below), or may present with sudden death as their initial symptoms.
Classification
• The estimated prevalence of CHD is 13,200,000, and one in three American adults is estimated to have some type of cardiovascular disease (CVD).1 Since 1900, CVD has been the number 1 cause of death every year except 1918.1 It is also the most costly medical condition in the United States.1
• ACSs are classified as unstable angina (UA), non-ST segment elevation myocardial infarction (NSTEMI) or ST segment elevation MI (STEMI) depending on serologic evidence of myocardial damage and on electrocardiographic (ECG) findings.
• Several classification schemes exist for grading stable angina, although the most commonly utilized may be the Canadian Cardiovascular Society system commonly referred to as CCSS.
Canadian Cardiovascular Society2 Class Description | |
I | Ordinary physical activity does not cause angina |
II | Slight limitation of ordinary physical activity |
III | Marked limitation of ordinary physical activity |
IV | Inability to perform any physical activity without discomfort |
• Most commonly in IHD, an obstruction develops due to atherosclerosis. Atherosclerotic plaques may be stable and result in a pattern of chronic angina precipitated when myocardial oxygen demand exceeds the supply that is available across a coronary artery obstruction. ACS typically occurs when an unstable coronary artery plaque ruptures, promoting thrombus formation on the surface and acutely obstructing the vessel lumen.
• The degree of coronary obstruction does not necessarily correlate with the likelihood of an acute coronary event; high-grade stenosis may never progress to infarction while less obstructive plaques may rupture and cause infarction. Some patients with angina have normal appearing coronary arteries. This situation can occur when the etiology of the symptoms is not cardiac, but it can also occur among patients who develop cardiac ischemia from coronary artery vasospasm (Prinzmetal angina). Those with Prinzmetal angina are typically younger and have fewer traditional IHD risk factors. The etiology is unclear and the prognosis (absent concomitant CAD) is favorable. Other patients may have microvascular disease or diffuse disease that is hard to detect on routine catheterization. These patients are more commonly women and may still have a substantial risk for progression to MI.
• Atherosclerosis is a chronic inflammatory disease resulting from a complex interplay between cellular and chemical factors affecting the vascular endothelium. The atherosclerotic process occurs over years; is triggered by traditional IHD risk factors such as smoking, obesity, diabetes, hypertension, hyperlipidemia, and genetics; and results in the formation of obstructing plaques. Investigation into other contributor factors continues, but the most recent evidence shows that 80% to 90% of patients with IHD have traditional risk factors3 and 87% to 100% of patients who suffer fatal IHD events have at least one traditional risk factor.4
• A number of novel serum markers have been found to be associated with heart disease. Some are risk factors and others are only markers of disease. High-sensitivity C-reactive protein (hs-CRP) has derived the most attention and appears to be an independent risk factor for heart disease. Lipoprotein-a is a nonmodifiable risk factor that is genetically programmed and signifies risk for early-onset heart disease. In contrast, elevated homocysteine elevates risk for heart disease, but lowering homocysteine levels does not reduce the risk of heart disease and is thus considered only a risk marker.
• IHD is associated with obstructive sleep apnea and snoring, but the nature and direction of this association are not yet clear.
DIAGNOSIS
Clinical Presentation
• An ACS may present with “typical” chest pain, atypical angina, sudden fatigue, congestive heart failure symptoms, or even nausea and vomiting. Diagnostic and management decisions must be made quickly and implemented immediately because the efficacy of many of the available treatments declines rapidly with time from onset of ischemia. Patients presenting with symptoms consistent with ACS should be classified very rapidly as having probable noncardiac pain, stable angina, UA/NSTEMI, or STEMI meeting reperfusion criteria.
• Stable angina commonly presents as exertional chest pain, tightness, shortness of breath, or fatigue. It has often been occurring for weeks or months before the patient consults a physician.
• IHD presents in enough ways and across a broad enough spectrum of patients that it would be misleading to describe a “typical” patient or presentation. Rather, a high index of suspicion should be maintained for IHD among men over 40 and women over 50, with a rapidly increasing prior probability with advancing age.
• Primary prevention of IHD is an important part of a primary care physician. Baseline risk among asymptomatic adults should be estimated using the National Cholesterol Education Program (NCEP) framework,5 either by counting risk factors or by calculating risk using the Framingham equation (accessible at http://cvdrisk.nhlbi.nih.gov/calculator.asp). The NCEP framework does overestimate risk among low-prevalence populations and can underestimate risk in some high-prevalence groups (e.g., Indian and other south Asian populations) by as much as 50%, so clinical judgment is required.
History
History is the most important information in the decision process for suspected IHD. It should address the following parameters:
• The location, character, and time course of the symptoms. Chest or left arm pressure or pain of a steady, dull nature is classic for cardiac ischemia. The feeling may be profound but vague and not even termed pain or pressure by the patient. Alternatively, some patients will insist that the sensation is one only of pressure, not pain. Occasionally pain may be present only in the jaw or scapular area. Sharp or pleuritic pain weighs against the diagnosis, as does pain that can be localized with one finger. Paresthesias (especially perioral tingling) suggest panic attack. Water brash has high specificity for gastroesophageal reflux. Reduced pain upon sitting up and leaning forward suggests pericarditis. Women may frequently present with vague symptoms that may be considered “atypical.”
• Prior history of IHD.
• Classic epidemiologic risk factors are smoking, hyperlipidemia, hypertension, obesity, and family history. These have little or no diagnostic value for ACS,6 but should be assessed in evaluating chronic angina. Those that are modifiable are key points in primary and secondary prevention.
• Diabetes. Patients with long-standing diabetes often lack the characteristic pain of acute ischemia.
• A complete listing of current medications including over-the-counter and alternative or herbal preparations and a list of any illicit drugs being used, especially cocaine.
A full cardiac and vascular review of systems should be included in the history for suspected chronic angina, but gathering this information should not be allowed to delay the rapid evaluation of ACS.
Physical Examination
• Physical examination in cases of suspected ACS should be expeditiously conducted and directed to key findings, which include pulmonary edema, particularly sudden or “flash” edema; mitral valve murmur, particularly if of new onset; marked hypertension; hypotension or shock; confusion or other mental status changes; other neurologic deficits consistent with stroke; and hypoxia. These findings can be detected by a careful assessment of the ABCs (airway, breathing, and circulation), review of vital signs, as well as a heart, lung, and focused neurologic examination.
• For patients with suspected chronic angina, a more complete directed physical examination emphasizing cardiovascular findings should be carried out. Carotid, abdominal, and renal bruits; pedal pulses (and ankle-brachial indices if pulses are diminished); and jugular venous waveform should be included. Chest tenderness to palpation that completely reproduces the presenting pain may make ACS less likely but does not exclude the possibility, and this finding must be interpreted within the context of other clinical data.
Laboratory Studies
• ECG is, with history, the foundation of diagnosis and risk stratification for suspected ACS. Certain crucial features are important:
• ST segment elevation of at least 1 mm in two contiguous leads
• New-onset left bundle branch block
• Either of the above findings on an ECG in a patient with chest pain is diagnostic of STEMI, and the patient should be triaged appropriately and emergently for reperfusion therapy via either thrombolytic therapy or emergent percutaneous coronary intervention (PCI). Patients with ST segment depression in the anterior leads in a pattern consistent with a posterior MI may also benefit from emergent reperfusion. Patients meeting these criteria must be identified immediately and emergently triaged from the outpatient setting to appropriate facilities to receive thrombolysis within 30 minutes or PCI within 90 minutes. Patients not meeting these criteria should not receive reperfusion therapy as it worsens outcomes.7
• Other ECG findings such as Q waves of 1 mm or greater not known to be present previously, T-wave inversion, hyperacute T waves (≥50% of the maximal QRS amplitude), ST segment depression, and new conduction abnormalities or arrhythmias may also be important markers of ischemia.
• Cardiac troponins T and I are more than 90% sensitive and similarly specific at 8 or more hours from the onset of pain.8 Either or both may be assayed, generally for levels >0.1 ng per mL. Positive troponins with normal ECG can distinguish NSTEMI from UA and help identify patients who are at increased risk for infarction or sudden death.
• All patients suspected of IHD should have a fasting lipid profile, glucose, complete blood count (CBC), estimated glomerular filtration rate (GFR), and electrolytes measured. Evaluation of ACS should not be delayed to obtain them; lipids may be measured fasting up to 24 hours after symptom onset.7
Imaging
• Patients suspected of IHD should have a chest radiograph performed for pulmonary edema and cardiac enlargement. Evaluation of patients with ACS and initiation of reperfusion therapy, however, should not be delayed for radiography.
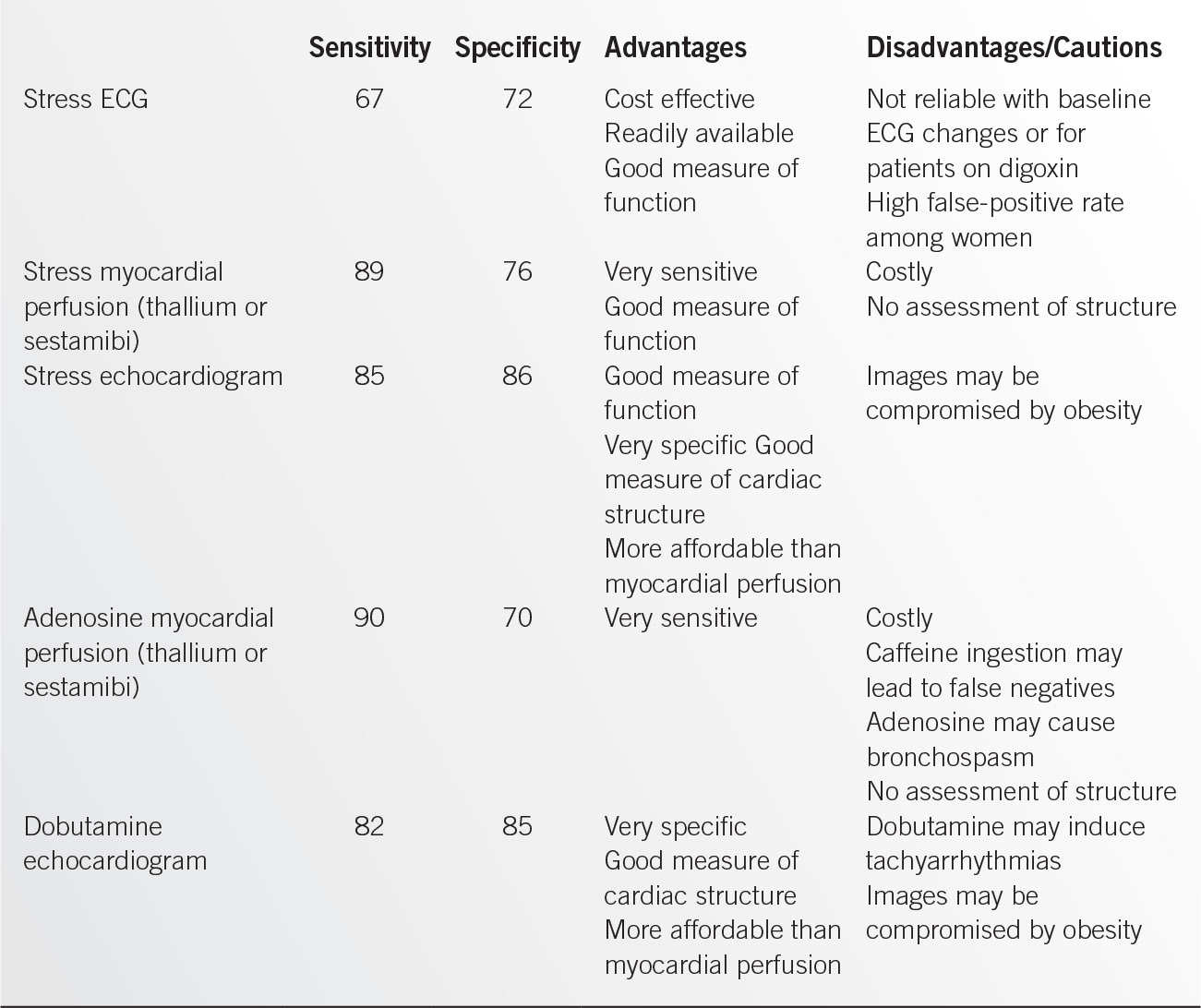
• Stress testing is employed to assess cardiac structure and function. It is commonly used to assess the probability of significant coronary artery disease among those who have anginal symptoms or who are preparing for noncardiac surgery. A number of different stress and imaging modalities are available. The advantages and disadvantages of the most commonly utilized forms are displayed in Table 9.2-1. If stress testing is to be used for this purpose, pretest probability needs to be considered before ordering an examination. If the pretest probability is very low, the result of a stress test may not influence decision making. If the risk is very high, then a negative test may not influence the decision whether a catheterization is necessary but may be helpful in targeting vessels for future revascularization.
• Stress testing may also be useful for assessing a patient’s functional capacity, assessing myocardial viability post-MI, and occasionally for guiding medical management of IHD.
• Left heart catheterization is the gold standard for evaluating coronary artery anatomy. It is generally safe and is appropriate for identifying the location and extent of obstructive disease. If suspicion for Prinzmetal or vasospastic angina is high, a heart catheterization may also be used with ergonovine to assess for the presence of coronary artery spasm.
• Computed tomography (CT) scan has evolved as an emerging technology that may have significant potential for heart disease. Multidetector CT and CT angiography are both being utilized and investigated, but the role that these emerging technologies will ultimately play in the diagnosis and management of heart disease is not yet clear.
Differential Diagnosis
• Patients presenting with chest pain and related complaints have ACS in a minority of cases, approximately 30% in the emergency department setting8 and less than 5% in the primary care physician’s office setting.10 Other high-probability diagnoses that should be considered are panic attack, gastroesophageal reflux disease, musculoskeletal pain, and pleurisy. Panic attack and gastroesophageal reflux disease are often close mimics of angina, and both are more common than angina in primary care settings. Both can result in morbidity, if misdiagnosed as angina, from inappropriate cardiac workups and from failure to treat the patient’s real condition. Also consider other life-threatening diagnoses such as aortic dissection, pulmonary embolus, pneumothorax, or perforating ulcer, among others.
TREATMENT
• Outpatient management of ACS focuses on rapid identification and risk stratification, immediate transport of reperfusion candidates to properly equipped facilities, and appropriate referral and transport of moderate- and high-risk patients. Patients without known true hypersensitivity or active bleeding should receive 325 mg of aspirin stat and be placed on 2 L per minute of oxygen by nasal cannula or mask while awaiting transport. If ECG monitoring is available, it should be in place. Nitroglycerine can be administered sublingual every 5 minutes as tolerated by blood pressure for pain relief. A defibrillator should be ready, and personnel trained in its use should be with the patient continuously. Time is of the essence in ACS. American College of Cardiology/American Heart Association (ACC/AHA) standards are to keep total ischemic time to less than 120 minutes.7 Patients with evidence of STEMI should be transported rapidly to a facility capable of providing appropriate reperfusion—thrombolysis or PCI.
• Outpatient management of IHD consists of primary and secondary prevention. Inpatient management of ACS is outside the scope of this chapter. Common to both primary and secondary intervention is risk factor reduction—weight loss; smoking cessation; and good control of hypertension, hyperlipidemia, and diabetes (in order of absolute benefit) if present. Risk factor reduction occurs through both behavioral and medical interventions. Treatment of hypertension should be to a BP goal of <140/90 for all patients including those with renal disease or diabetes. Lipid lowering should be achieved with a statin, if tolerated, and in conjunction with the NCEP goals as described below.5 National treatment goals for diabetes are for a HbA1c <7.0, though available evidence does not clearly demonstrate reduced ACS risk for such tight diabetes control.
Population | Goal |
<2 risk factors | Low-density lipoprotein (LDL) <160 mg/dL |
2 or more risk factors | LDL <130 mg/dL |
Known IHD or diabetes | LDL <100 mg/dL |
Very high-risk patients | Optional LDL target of 70 mg/dL (recent ACS, IHD plus diabetes; metabolic syndrome) |
• Operative interventions, for example, coronary artery bypass grafting (CABG) or percutaneous intervention (PCI) with stent placement, are available for treatment of disease in appropriate patients.
Behavioral
• Smoking is the most powerful modifiable risk factor for IHD, and smoking cessation is essential for both primary and secondary prevention. Physicians should ask about smoking habits at each visit, counsel the patient to quit, assess the willingness to quit, and assist the patient in quitting smoking.
• Depression is thought to be an independent risk factor for IHD and ACS and is associated with a worse prognosis. Treatment with selective serotonin-reuptake inhibitors (SSRIs) improves depression morbidity though not cardiac outcomes.11 Current expert opinion is to use SSRIs among patients with IHD as needed in a manner consistent with how they would be used in the absence of heart disease.
• Weight loss is an important component of reducing risk for MI. Body mass index (BMI) has traditionally been used as a measure of risk with the goal being <25, but waist-to-hip ratio has emerged as a more reliable measure of risk. Weight loss should target a waist circumference of <40 inches in men and <35 inches in women.12
• Exercise both reduces risk directly and is an important component of weight loss. A written exercise prescription should be given for 30 to 60 minutes of activity, defined as brisk walking, 5 to 7 days per week,12 and progress toward that goal should be monitored and reinforced at every visit. Patients may opt for more vigorous activity based on stress test results.
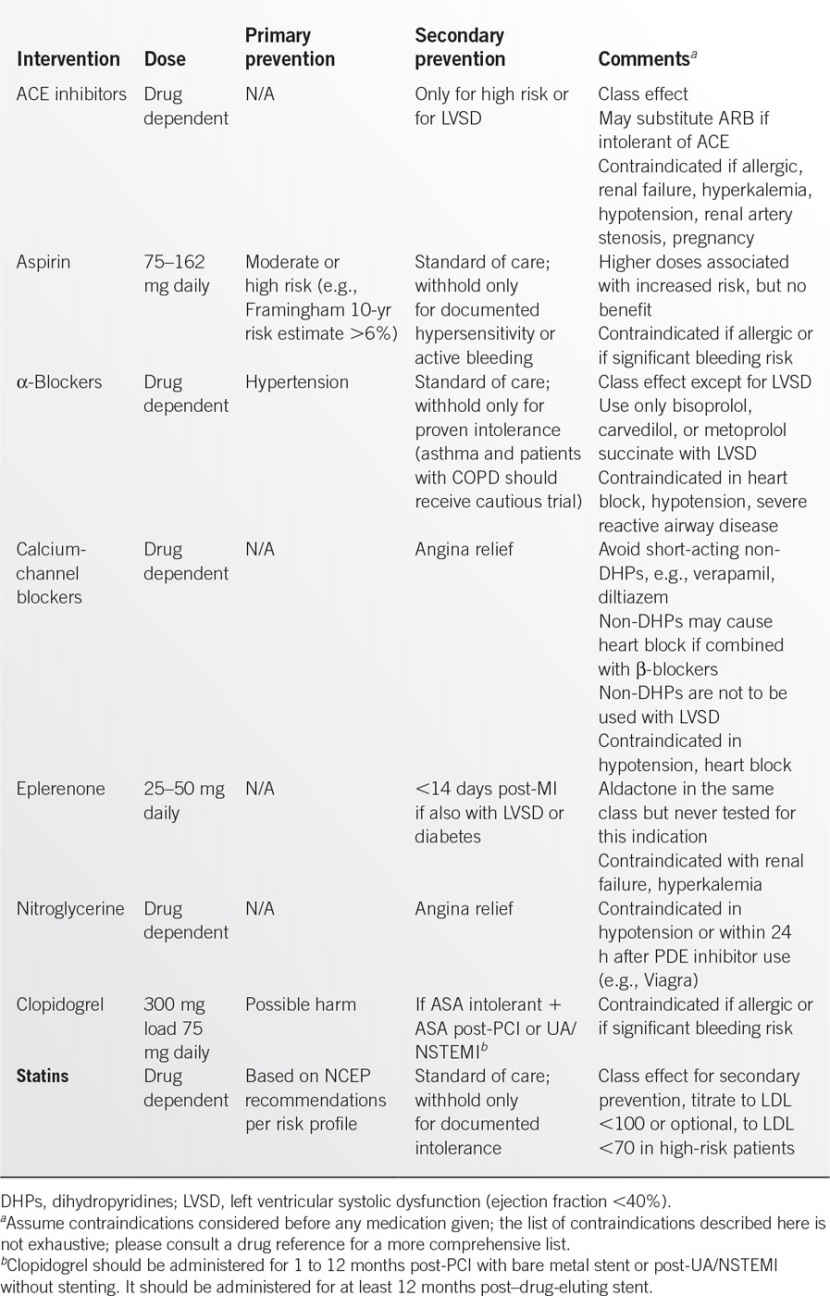
Medications
• Medical management of IHD can involve a diverse array of medications. The most common are described in Table 9.2-2. The three medications in bold type are recommended to all patients unless contraindicated, with regular assessment of compliance. Intensive medical therapy is capable of achieving regression of coronary plaques and reducing ACS events.
Surgery
• Emergent reperfusion therapy reduces mortality and morbidity, and is the standard of care for STEMI meeting the criteria above. PCI is preferred in high-volume centers if door-to-balloon times of 90 minutes or less can be achieved. Thrombolysis should be initiated (with a target door-to-needle time of 30 minutes) if suitably skilled PCI is not available in that time frame.13 The primary care physicians must honestly assess the procedure volume and skills of their referral facility and the realistically likely time to initiation of therapy in making referral decisions.
• Among stable angina patients, CABG improves survival for patients with left main disease, severe proximal left anterior descending (LAD) disease, or three-vessel disease with diminished left ventricular (LV) function. (A vessel is considered diseased if it has ≥50% obstruction on coronary angiography.) Recent improvements in angioplasty technology, particularly stenting, may make percutaneous revascularization an appropriate alternative for some such patients. Patients with diabetes do not fare as well with percutaneous revascularization as with CABG.
• Anginal pain, ability to exercise, and daily role function are important patient-oriented outcomes. Consultation and evaluation for revascularization (either percutaneous or by CABG) to reduce pain and improve function is appropriate for many patients with stable IHD, even if mortality is unlikely to be reduced.
Special Therapy
• Yearly influenza vaccine is indicated among patients with IHD. Also, patients with IHD should have a pneumovax once, which is to be repeated when the patient is older than 65 if the first pneumovax was administered before the patient was 65 years old.
SPECIAL CONSIDERATIONS
• The prevalence and the magnitude of impact of IHD have made appropriate management of this condition a high priority among groups following the quality of care for chronic diseases. Physicians should follow quality standards such as aspirin, statins, and β-blockers for secondary prevention and should develop systems for identifying and tracking patients with IHD.
REFERENCES
1. Thom T, Haase N, Rosamond W, et al. Heart disease and stroke statistics 2006 update. A Report from the American Heart Association Statistics Committee and Stroke Statistics Subcommittee. Circulation 2006;113:e85–e151.
2. Goldman L, Hashimoto B, Cook EF, et al. Comparative reproducibility and validity of systems for assessing cardiovascular functional class: advantages of a new specific activity scale. Circulation 1981;64:1227–1234.
3. Khot UN, Khot MB, Bajzer CT, et al. Prevalence of conventional risk factors in patients with coronary heart disease. JAMA 2003;290(7):898–904.
4. Greenland P, Knoll MD, Stamler J, et al. Major risk factors as antecedents of fatal and nonfatal coronary heart disease events. JAMA 2003;290(7):891–897.
5. National Cholesterol Education Program. National Heart, Lung, and Blood Institute. http://www.nhlbi.nih.gov/guidelines/cholesterol/atp_iii.htm. Accessed June 30, 2006.
6. Jayes RL, Beshansky JR, D’Agostino RB, et al. Do patients’ coronary risk factor reports predict acute cardiac ischemia in the emergency department? A multicenter study. J Clin Epidemiol 1992;45:621.
7. Antman EM, Anbe DT, Armstrong PW, et al. ACC/AHA guidelines for the management of patients with ST-elevation myocardial infarction; a report of the American College of Cardiology/American Heart Association Task Force on Practice Guidelines (Committee to Revise the 1999 Guidelines for the Management of Patients with Acute Myocardial Infarction). J Am Coll Cardiol 2004;44(3):E1–E211.
8. Ebell MH, Flewelling D, Flynn CA. A systematic review of troponin T and I for diagnosing acute myocardial infarction. J Fam Pract 2000;49:550.
9. Murthy TH, Bach DS. Comparative review of stress tests. Clin Fam Pract 2001;3(4):814.
10. Klinkman MS. Episodes of care for chest pain. J Fam Pract 1994;38:345.
11. Agency for Healthcare Research and Quality. Post-myocardial infarction depression (Evidence report/technology assessment report 123). Rockville, MD: U.S. Government Printing Office; 2005. AHRQ publication 05-E018-02.
12. Smith SC, Allen J, Blair SN, et al. AHA/ACC guidelines for secondary prevention for patients with coronary and other atherosclerotic vascular disease: 2006 update. Circulation 2006;113:2363–2372.
13. Van de Werf F, Gore JM, Avezum A, et al; GRACE Investigators. Access to catheterisation facilities in patients admitted with acute coronary syndrome: multinational registry study. BMJ 2005;330:441–444.
| Murmurs and Valvular Heart Disease |
GENERAL PRINCIPLES
A heart murmur may have no pathologic significance—simply a representation of physiologic increases in blood flow. However, a murmur may be an important indicator of the presence of valvular abnormalities. The history and physical examination is the critical screening tool for all patients. In certain instances, further evaluation with electrocardiogram, chest x-ray, echocardiogram, and heart catheterization is required. Diagnosis is important in valvular heart disease in order to achieve timely management prior to the onset of irreversible damage. Timing of surgical intervention correlates with good outcome. Generally, patients with stenotic valvular lesions can be monitored clinically until symptoms appear. On the other hand, patients with regurgitant valvular lesions require careful echocardiographic monitoring for left ventricular function and may require surgery even in the absence of symptoms. A brief discussion of some basic diagnostic tools is listed below. In addition, the most common valvular heart diseases as well as murmurs in pregnancy, murmurs in athletes, and murmurs in infants and children are reviewed in the following text.
DIAGNOSIS
History
History suggesting valvular heart disease is directed at symptoms potentially related to dysfunction of a valve. These symptoms can be thought of as relating to diminished forward flow (fatigue and decreased exercise tolerance) and symptoms relating to pulmonary congestion (paroxysmal nocturnal dyspnea and orthopnea).
Physical Examination
The physical examination focuses on the location, timing, duration, and quality of the murmur. In addition to these cardinal elements, various provocative maneuvers can cause changes in the murmur, changes that aid diagnosis. The Valsalva maneuver and standing decrease preload. Squatting or raising the legs increases preload. Handgrip increases afterload. There is no maneuver that decreases afterload. The beat after the long pause associated with a premature beat may also give clues to the etiology of a murmur by causing increased filling of the left ventricle.
On cardiac physical examinations, murmurs need to be described and characterized to predict prompt management; for this reason, murmurs needs to be described as below.
Murmur description: A murmur is described by different number of features, including intensity (grade), frequency, timing, shape, location, and radiation.
Intensity: The intensity of a murmur is primarily determined by the quantity and velocity of blood flow at the site of its origin, the transmission characteristic of the tissues between the blood flow and stethoscope, the site of auscultation or recording, and the distance of transmission. In general, the intensity declines in the presence of obesity, emphysema, and pericardial effusion.
Murmurs are usually louder in children and in thin individuals.
Six grades are used to classify the intensity of a murmur:
• Grade I is the faintest murmur that can be heard (with difficulty)
• Grade II murmur is also a faint murmur but can be identified immediately
• Grade III murmur is moderately loud
• Grade IV murmur is loud and is associated with a palpable thrill
• Grade V murmur is very loud, could be heard placing the edge of the diaphragm of stethoscope over the patient’s chest, and is associated with a palpable thrill
• Grade VI murmur is the loudest and can be heard without a stethoscope
Pitch: The frequency of the murmur determines the pitch, which may be high or low. It can be described as harsh, rumbling, scratchy, grunting, blowing, squeaky, and musical. Quality and pitch are closely related.
Configuration: The time course of murmur intensity corresponds to the “shape” of a diagram of murmur intensity over time, as in a phonocardiogram. A number of configurations or shapes of murmurs are recognized:
• Crescendo
• Decrescendo
• Crescendo–decrescendo (diamond shaped)
• Plateau (unchanged in intensity)
Location: The location on the patient’s chest where the murmur is loudest is typically described as apical or parasternal. Parasternal murmurs are further described by the intercostal space and right or left side of the sternum.
Timing: The duration of a murmur is assessed by determining the length of systole or diastole that the murmur occupies. The murmur can be long (e.g., it occupies most of systole or diastole) or brief. The following classification is useful2:
• For systolic murmurs:
• Midsystolic (or systolic ejection)
• Holosystolic (or pansystolic)
• Early systolic
• Late systolic
• For diastolic murmurs:
• Early diastolic
• Mid-diastolic
• Late diastolic (or presystolic)
Laboratory Studies
• Electrocardiogram (ECG). The ECG is not a specific tool for the diagnosis of valvular heart disease. Findings such as atrial enlargement or left ventricular hypertrophy (LVH) often occur late in the course of valvular heart disease.
• Chest x-ray (CXR). Like the ECG, the CXR does not offer early or specific diagnostic clues to valvular heart disease. Radiographic evidence of cardiomegaly or pulmonary congestion is a late finding.
• Echocardiogram. The echocardiogram is the definitive indicator that rules in or rules out the presence of valvular heart disease. It should be used when there is moderate clinical suspicion of valvular heart disease.
SPECIFIC DIAGNOSIS AND TREATMENT BASED ON VALVULAR DISEASE OR CONDITION
Aortic Stenosis (AS)
General Principles
Pathophysiology. Left ventricular outflow obstruction leads to increased left ventricular pressure. In order to maintain normal wall stress, the left ventricle undergoes concentric hypertrophy. Subsequently, a decrease in contractile performance and in ejection fraction is noted.
Etiology of Valvular AS. Senile AS (age-related degenerative calcific changes), congenitally bicuspid vale with superimposed calcification, rheumatic heart disease.
Diagnosis
Clinical presentation. Exertional dyspnea, angina pectoris, syncope, congestive heart failure, and sudden death.
Physical Examination
• Murmur: Harsh, diamond-shaped systolic murmur. AS murmur is heard best in second right intercostal space and radiates into neck vessels. It gets softer with maneuvers that increase afterload (handgrip).
• Diminished intensity (or absence) of aortic valve closure
• Weakened (parvus) and delayed (tardus) upstroke of carotid artery pulsation
• Narrow pulse pressure
Treatment
Management. Asymptomatic AS management includes close clinical follow-up to monitor aortic valve area (normal is 3 to 4 cm2). In addition, patients require endocarditis antibiotic prophylaxis and avoidance of medication that could result in hypotension. Symptoms occur late in the course of disease and are an ominous sign. Onset of symptoms triggers the need for surgical evaluation.
Surgery. Aortic valve replacement is indicated if the patient becomes symptomatic, if there is evidence of left ventricular dysfunction, or if the patient has an expanding poststenotic aortic root. Percutaneous balloon aortic valvuloplasty is preferable in children and young adults with congenital, noncalcific AS.
Special Considerations: Subvalvular Aortic Stenosis
Hypertrophic cardiomyopathy (with outflow obstruction). This is a familial disease characterized by marked hypertrophy of the left ventricle, most commonly the interventricular septum. The murmur is similar to valvular AS, but differs in that any maneuver that will make the left ventricle larger in diastole with make the subvalvular AS murmur softer. Conversely, any maneuver that will decrease the left ventricular size in diastole will make the murmur louder. This is the most common cardiac abnormality found in young athletes who die suddenly during vigorous physical activity.
• Special therapy: β-Blockers are the standard of therapy, whereas calcium-channel blockers are sometimes useful.
• The guidelines for surgical intervention (myomectomy) are not well defined.
• The incidence of sudden death is 2% to 4% per year in adults and 4% to 6% per year in children and adolescence.
Mitral Stenosis
General Principles
Pathophysiology. Thickening and immobility of the mitral valve leaflets cause obstruction of blood flow from the left atrium to left ventricle and increased pressure within the left atrium, pulmonary vasculature, and right heart. A decreased mitral valve orifice (normal 4 to 6 cm2) requires an abnormally elevated left atrioventricular pressure gradient to move blood from the left atrium to the left ventricle. The elevated pulmonary venous and pulmonary arterial wedge pressures reduce pulmonary compliance, contributing to clinical symptoms.
Etiology. Mitral stenosis (MS) and mixed MS and mitral regurgitation (MR) are generally rheumatic in origin. Other etiologies include infective endocarditis and mitral annular calcifications. Rarely, congenital defects, endomyocardial fibroelastosis, malignant carcinoid syndrome, and systemic lupus erythematosis cause MS.
Diagnosis
Clinical presentation. Many patients deny symptoms because patients gradually reduce activity with the slow progression of disease. Clinical presentation includes:
• Exertional dyspnea (most common and often only symptom)
• Hemoptysis
• Thromboembolism
• Chest pain
• Infective endocarditis
• Right-sided heart failure
Physical Examination
• Murmur: Low-pitched, rumbling, diastolic murmur, heard best at the apex with the patient in the left lateral decubitus position. (Duration of the murmur corresponds with the severity.)
• Accentuated S1
• Opening snap
• Prominent “a” wave in jugular venous pulsations with normal sinus rhythm
Treatment
Management. An annual history and physical examination, as well as a CXR and ECG, are recommended in asymptomatic patients. Endocarditis prophylaxis is indicated in patients with MS; however, no further medical therapy is indicated. When mild symptoms develop, diuretics may be helpful in reducing left atrial pressure and decreasing symptoms. If symptoms are more than mild or if there is evidence of pulmonary hypertension, mechanical intervention is warranted and delaying intervention worsens prognosis.
Surgery. Mitral balloon valvotomy is indicated in symptomatic patients with isolated MS whose valve orifice is <1.7 cm2. Balloon valvotomy is the procedure of choice in individuals with mobile, thin leaflets with no or little calcium. If balloon valvotomy is not possible, a surgical (“open”) valvotomy can be performed. Mitral valve replacement is indicated in individuals with MS and significant associated MR.
Aortic Regurgitation
General Principles
Etiology
• Abnormalities of valve leaflets: Rheumatic heart disease, endocarditis, congenital
• Aortic root disease: Aortic dilation/dissection, syphilitic aortitis, Marfan syndrome, rheumatoid spondylitis
Pathophysiology. In AS, an abnormal regurgitation of blood from the aorta to the left ventricle occurs during diastole. As a result, the left ventricle must pump the regurgitant volume in addition to the normal volume returning from the left atria. An increase in left ventricular end-diastolic volume is the main hemodynamic compensation. The left ventricle undergoes adaptive change, namely dilation and eccentric hypertrophy.
Diagnosis
Clinical presentation. Symptoms of dyspnea on exertion, fatigue, and decreased exercise tolerance appear due to left ventricular failure. Also, patients with AR may experience an uncomfortable sensation associated with large pulse pressure.
Physical Examination
• Murmur: Blowing diastolic murmur which is best heard with the patient leaning forward, after exhaling. The murmur may get louder with increased afterload (handgrip).
• Bounding pulse
• Widened pulse pressure
• Displaced cardiac impulse (down and to patient’s left)
Treatment
• Management. Asymptomatic patients require regular clinical evaluation, assessment of left ventricular function, and endocarditis antibiotic prophylaxis. The mainstays of medical management in symptomatic patients are afterload reduction (vasodilators), which reduces the amount of aortic regurgitations. Long-acting nifedipine has been shown to delay the need for valve surgery.
• Surgery. Compelling evidence supports surgical correction before the onset of permanent left ventricular damage, even in asymptomatic patients. AR should be corrected in patients who remain symptomatic despite optimal medical therapy. Aortic valve replacement should also be performed with progressive left ventricular dysfunction and a left ventricular ejection fraction <55% or left ventricular end-systolic volume >55%—“55/55 Rule” (even if asymptomatic).
Mitral Regurgitation
General Principles
• Pathophysiology A portion of the left ventricular output is forced backward into the left atrium (LA) leaving the forward cardiac output into the aorta reduced. In acute MR, the LA is normal size and relatively noncompliant. LA pressure rises dramatically with subsequent pulmonary edema and right heart failure. In chronic MR, dilation and eccentric hypertrophy of the LA occur, making the LA more compliant; therefore, pulmonary edema is less likely to develop.
Etiology
• Acute MR: Endocarditis, ruptured chordae, papillary muscle dysfunction
• Chronic MR: Rheumatic heart disease, myxomatous degeneration, congenial anomaly, infective endocarditis, hypertrophic cardiomyopathy
Diagnosis
Clinical presentation. The most common symptoms with chronic, severe MR include fatigue, exertional dyspnea, and orthopnea. Patients with pulmonary vascular disease can develop right-sided heart failure. In acute, severe MR, left ventricular failure with acute pulmonary edema is common.
Physical Examination
• Murmur: Apical, holosystolic murmur at apex with radiation to left axilla. The murmur of MR will become louder with increased afterload (handgrip).
• Presence of S3, which indicates severe disease
• Laterally displaced cardiac impulse
Treatment
• Management. Asymptomatic patients require regular clinical evaluation, assessment of left ventricular function, and endocarditis antibiotic prophylaxis. In a normotensive patient with acute severe MR, nitroprusside can be utilized to diminish the amount of MR, in turn increasing forward output and reducing pulmonary congestion. For the asymptomatic patient with chronic MR, there is no generally accepted medical therapy. There are no large long-term studies to indicate that the use of vasodilators are beneficial in chronic MR. Heart rate should be controlled with digitalis, rate-lowering calcium-channel blockers, or β-blockers if atrial fibrillation develops.
• Surgery. The optimal timing of surgery in patient with chronic MR can be a difficult decision. Routine echocardiographic evaluation should be performed in individuals with severe MR. Surgery is recommended when a patient is symptomatic despite optimum medical management. Surgery should also be considered when left ventricular dysfunction is progressive, with left ventricular ejection fraction declining below 60% (even if asymptomatic).
Special Considerations
• Mitral valve prolapse (MVP). MVP is an exceedingly common condition and often asymptomatic. Patients may present with symptomatic arrhythmia, atypical chest pain, or exaggerated autonomic symptoms. Physical examination reveals a click (with or without a murmur), which move toward S2 with increased preload and increased afterload. Some patients require endocarditis antibiotic prophylaxis. The degree of pathology is related to the degree of MR. β-Blockers can be used for symptomatic treatment of chest pain.
Valvular Heart Disease in the Athlete
Preparticipation Physical
The preparticipation physical should focus on a family history of heart disease; sudden death; personal history suggesting syncope, near syncope, or arrhythmia; and evaluation of heart murmurs in supine, sitting, standing, squatting, and postsquatting positions.
High-Risk Murmurs
Most common causes of serious valvular heart disease in athletes causing sudden death are mitral prolapse and subaortic stenosis (caused by hypertrophic cardiomyopathy).
Risk Assessment
The main issue with MVP is the degree of ectopy present, especially with exercise. In hypertrophic cardiomyopathy, the most significant problem is the degree of outflow obstruction, which is usually related to the thickness of the septum.
Valvular Heart Disease in Pregnancy
Etiology
Most murmurs in pregnancy are physiologic as there is a 50% increase in circulating blood volume during pregnancy.
Preexisting Disease
Preexisting valvular heart disease often is exacerbated by pregnancy. The increased blood volume and enhanced cardiac output associated with normal pregnancy can accentuate the murmurs associated with stenotic heart valve lesions (e.g., MS, AS), whereas murmurs of AR or MR may actually ease in the face of lowered systemic vascular resistance.
Valvular Lesions With Increased Maternal and Fetal Risk
• Severe AS with or without symptoms
• MR or AR with NYHA functional Class III to IV symptoms
• MS with NYHA functional Class II to IV symptoms
• Valve disease resulting in severe pulmonary hypertension (pulmonary pressure >75% of systemic pressures)
• Valve disease with severe left ventricular dysfunction (EF <0.40)
• Mechanical prosthetic valves requiring anticoagulation
• AR in Marfan syndrome
Valvular Heart Disease in Infants and Children
Etiology
The physician must consider valvular heart disease as a subset of congenital heart disease. In diagnosis of murmurs in infants and children, think of congenital problems and then rule in or out a valvular etiology.
• Left to right shunts, for example, ventricular septal defect (VSD) or atrial septal defect (ASD)
• Obstructive lesions, such as AS, pulmonic stenosis, coarctation of the aorta
• Valvular insufficiency
Relative frequency of pathologic murmurs in infants: Of murmurs in congenital heart disease, 63% are caused by the six most common congenital defects:
• Pulmonic stenosis > PDA > ASD > Coarctation of the aorta > Aortic stenosis
Diagnosis
Findings more common in infants and children than in adults include grunting, poor feeding, sweating, poor weight gain, wheezing, decreased exercise tolerance, cough, and squatting after exercise (to increase preload). Cyanosis and edema are very late findings.
Treatment
• Referrals. Pediatric cardiologists do not order echocardiograms in a large percentage of patients seen in referral for murmur. This makes the strategy of referring all questionable murmurs to a pediatric cardiologist more cost-effective than ordering echocardiograms and referring only the pediatric patients with positive findings on echo.
• Surgery. Children who have congenital heart disease that might require surgery should be treated with input from a pediatric cardiologist. Reasons not to operate include the fact that some structural problems, such as VSD and PDA, sometimes resolve on their own. Other reasons not to operate include the fact that younger children are poorer operative candidates and that artificial valves will need to be replaced as the child grows. Reasons not to wait too long include irreversible processes (such as pulmonary hypertension) and irreversible structural damage (such as dilatation or hypertrophy of the ventricles).
REFERENCES
1. Bonow RO, Carabello B, de Leon AC, et al. Guidelines for the management of patients with valvular heart disease: executive summary. A report of the American College of Cardiology/American Heart Association Task Force on Practice Guidelines (Committee on Management of Patients with Valvular Heart Disease). Circulation 1998;98:1949–1984.
2. Boon NA, Bloomfield P. The medical management of valvular heart disease. Heart 2002;87:395–400.
3. Carabello BA, Crawford FA. Valvular heart disease. N Engl J Med 1997;337:32–41.
4. Davies MK, Gibbs CR, Lipp GYH. ABC of heart failure—investigation. BMJ 2000;2730:297–300.
5. Liberthson RR. Sudden death from cardiac causes in children and young adults. N Engl J Med 1996;334:1039–1044.
6. Rosenhek R, Binder T, Porenta G, et al. Predictors of outcome in severe asymptomatic aortic stenosis. N Engl J Med 2000;343:611–617.
7. Scognamiglio R, Rahimtoola SH, Fasoli G, et al. Nifedipine in asymptomatic patients with severe aortic regurgitation and normal left ventricular function. N Engl J Med 1994;331:689–694.
8. Shipton B, Wahba H. Valvular heart disease: review and update. Am Fam Physician 2001;63:2201–2208.
9. Spirito P, Bellone P, Harris K, et al. Magnitude of left ventricular hypertrophy and risk of sudden death in hypertrophic cardiomyopathy. N Engl J Med 2000;342:1778–1785.
10. Stapleton JF. Natural history of chronic valvular heart disease. Cardiovasc Clin North Am 1986;16:105–149.
11. Chatterjee K. Auscultation of Heart Murmurs. Up to date. http://www.uptodate.com/contents/auscultation-of-cardiac-murmurs. Accessed April 23, 2014.
|
GENERAL PRINCIPLES
Definition
Heart failure (HF) is a complex clinical syndrome deriving from cardiac dysfunction that may be either acute or chronic in its presentation. The “classic” presentation of acute, severe HF evokes an image of a dyspneic patient sitting upright due to pulmonary edema with poor peripheral perfusion. However, identical or more severe hemodynamic abnormalities are commonly found in the patient with chronic HF without extreme symptoms or dramatic physical examination signs, reflecting a slower, insidious onset. Therefore, while acute HF is readily diagnosed, signs and symptoms of chronic HF are frequently overlooked in clinical practice.
The syndrome of HF can result from congenital or acquired abnormalities of cardiac muscle (endocardium, myocardium) or from valvular, great vessel, or pericardial disorders. HF results when the heart is unable to generate cardiac output sufficient to meet and maintain the metabolic requirements of the body without a marked elevation in filling pressure (at rest, upon exertion, or under other physiologic demands). This chapter focuses on the diagnosis, evaluation, and treatment of the patient with chronic HF due to left ventricular (LV) dysfunction. This working definition of chronic HF still does not identify disease physiology in terms of the degree of systolic (ejection-related) or diastolic (relaxation-related) LV dysfunction, nor infer disease etiology. Chronic HF may be associated with a wide spectrum of LV functional abnormalities, which may range from patients with normal LV size and preserved ejection fraction (EF) to those with severe LV dilatation and/or markedly reduced EF. In most patients, abnormalities of systolic and diastolic dysfunction coexist, regardless of LV EF.
Patients with an LV EF ≤40% are classified as having heart failure with reduced EF (HF-rEF), whereas those with EF ≥50% are classified as heart failure with preserved EF (HF-pEF). Patients with HF-pEF are further classified as borderline HF-pEF if EF is 41% to 49% or improved HF-pEF if EF has improved to greater than 40%, respectively.1 The latter two categories represent a heterogeneous and intermediate group of patients in whom the optimal treatment and clinical outcomes are undefined and understudied.
Epidemiology
HF is an invariably progressive syndrome affecting over 5 million persons in the United States. It is the only cardiovascular disorder with increasing prevalence, especially among elderly individuals and in women. More than 550,000 new cases of HF are diagnosed annually, and both HF-pEF and HF-rEF appear equal in frequency. The incidence of HF approaches 10 per 1,000 population after age 65 and approximately 80% of patients hospitalized with HF are more than 65 years old.1–4 By the year 2050, one in five Americans will be over 65 years of age.4 HF is the primary diagnosis in more than 1 million hospitalizations annually.3 Patients hospitalized for HF are at high risk for recurrent hospitalizations, with a 1-month readmission rate of 25%.2
At the age of 40 years, the lifetime risk of developing HF in Americans is 20%.3 This common, yet generally preventable, syndrome is characterized by high mortality, frequent hospitalization, and reduced quality of life. It is the most common Medicare diagnosis-related group, and more Medicare dollars are spent for the diagnosis and treatment of HF than for any other diagnosis. The total cost of HF care in the United States exceeds $30 billion annually, with over half of these costs spent on hospitalizations.2–4 Despite marked advances in medical and surgical therapy over the past two decades, the morbidity and mortality from HF remain unacceptably high, averaging 10% mortality at 1 year and 50% mortality at 5 years.3
Pathophysiology
HF commonly results from a single acute event, or due to chronic or repetitive cardiac injury. Inciting factors include conditions as disparate as myocardial infarction (MI) and myocardial damage due to viral myocarditis, alcohol, or a chemotherapeutic agent. This can be explained by the observation that regardless of the nature of the cardiac injury, the adaptive systemic response to altered cardiac function and hemodynamics as well as the resultant cardiac structural changes and cellular processes that develop within the heart itself are remarkably consistent. The characteristic pathophysiology of HF derives from systemic and local cardiac neurohormonal activation designed to be compensatory in nature, but results in deleterious changes in myocardial structure and cellular function in areas that were previously normal.5 This process is termed “cardiac remodeling,” whose key features include the following:
• Remodeling is initiated by a threshold-reaching injury to the heart, resulting in systemic and local neurohormonal activation—renin–angiotensin–aldosterone (RAAS) and sympathetic nervous systems.
• Neurohormonal activation results in additional myocardial damage that continues after resolution of the initiating event and tends to progress over time.
• Cardiac remodeling therefore results in increased cardiac chamber volumes and muscle mass (eccentric LV hypertrophy), increased extracellular matrix deposition, and myocardial fibrosis.
Etiology
The most common etiology of HF in the United States is ischemic heart disease.1 Hypertensive or valvular heart disease and primary cardiomyopathy (familial or idiopathic) are also common. Myocardial dysfunction can be secondary to infectious, metabolic, endocrine, nutritional, or toxic causes (notably alcohol and anthracyclines); acute stress (Takotsubo cardiomyopathy), connective tissue or pericardial diseases; neuromuscular or autoimmune disorders; as well as infiltrative diseases (amyloidosis, iron overload, sarcoidosis) or undiagnosed congenital heart disease. This chapter will not address the category of high-output HF (due to thyrotoxicosis, sepsis, severe anemia, beriberi, Paget disease, myeloma, pregnancy, or significant arteriovenous shunting).
Classification
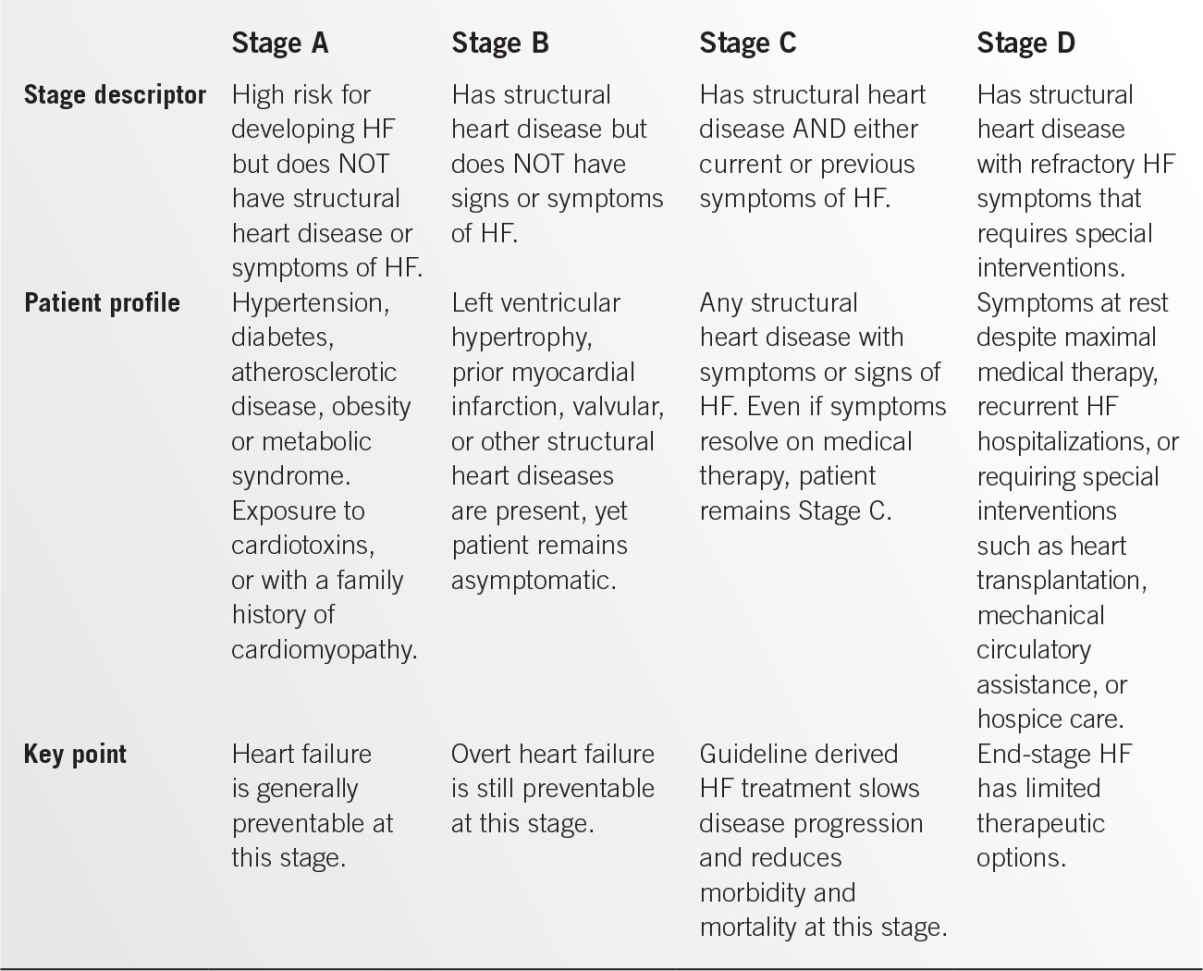
The American College of Cardiology and the American Heart Association adopted an innovative approach to the classification of HF beginning in 2001. This classification scheme emphasizes risk factors for both the development and progression of the disease.1 Four well-defined stages comprise the HF syndrome. The first two stages (A and B) were devised to assist health care providers to more easily identify patients at risk for developing HF with the goal of disease prevention in mind. Stage C denotes the majority of patients who have been diagnosed with clinical HF, and Stage D denotes patients who have developed refractory HF despite optimal therapy (Table 9.4-1).
Within Stage C, the New York Heart Association (NYHA) classification system is traditionally employed to categorize HF symptoms and estimate prognosis in clinical trials. To be useful in practice, one must consider the patients’ baseline subjective symptoms in reference to a normal or expected activity level for someone their age. The NYHA Symptom classification is as follows:
• Class I patients have no perceived symptoms or limitations in performing ordinary physical activities.
• Class II patients have symptoms of HF with slight or moderate levels of physical activity.
• Class III patients have a marked limitation of exercise tolerance, symptoms with simple activities of daily living but remain comfortable at rest.
• Class IV patients have symptoms of HF at rest.
DIAGNOSIS
Clinical Presentation
The clinical presentation of a patient with HF can be subtle, and patients with significant degrees of LV dysfunction may remain asymptomatic for some time. Because early diagnosis and treatment reduce morbidity and mortality, successful therapy depends on a high level of clinical suspicion and screening for signs and/or symptoms of HF in all patients at risk for its development (Stages A and B).1 The clinical presentation of a patient with Stage C (overt) HF may be acute but often is more insidious and progressive. Acute or sudden-onset HF symptoms (minutes to hours) should prompt evaluation for myocardial ischemia/infarction, arrhythmia, acute valvular or LV structural deterioration, or hypertensive urgency producing a rapid, abrupt change in LV pressure or volume-loading conditions. Slow or gradual onset HF symptoms (days to weeks) is more common, as mild symptoms of HF are often unrecognized or ignored by the patient until they become severe or persistent at rest.
History and Symptoms
• Pertinent historical elements should include information regarding the risk factors for HF, the type and/or the extent of cardiac structural abnormalities present and the temporal nature or duration of the cardiac injury.6 Risk factors include hypertension, diabetes, dyslipidemia, coronary or peripheral vascular disease, skeletal or cardiac myopathy, valvular heart disease, rheumatic fever, mediastinal irradiation, sleep-disordered breathing, exposure to cardiotoxic agents, current/past alcohol, cocaine or amphetamine abuse, smoking, collagen vascular diseases, HIV infection, thyroid or other metabolic disorders, pheochromocytoma, other systemic diseases (e.g., sarcoidosis, amyloidosis, hemosiderosis) and morbid obesity. In addition, a family history of sudden cardiac death, cardiomyopathy, or tachyarrhythmia should be sought. If familial cardiomyopathy is suspected, a more detailed family history should be obtained, preferably including three generations.1
• Symptoms strongly suggesting a diagnosis of HF include dyspnea at rest or with exertion, orthopnea, paroxysmal nocturnal dyspnea (PND), nocturnal or recumbent cough or other sleep disturbance, pedal or scrotal swelling, impaired exercise capacity or endurance. Less specific presentations of HF include early satiety, nausea and vomiting, abdominal discomfort or bloating, exertional wheezing, unexplained fatigue, weakness, or malaise, mental confusion or impaired concentrating ability, and daytime oliguria with recumbent nocturia. The spectrum of symptoms in a given patient reflects the relative extent of systemic and/or pulmonary venous congestion related to fluid overload versus reduced cardiac output (hypoperfusion).
• In a patient with known LV dysfunction and previously diagnosed HF, provocative and exacerbating factors should be reviewed.1 Serial monitoring of weight gain or loss, medication and diet adherence, appetite, activity tolerance and sleep quality may reveal pitfalls to the most optimal therapeutic plan. Common precipitants of decompensation are excess dietary sodium, medication noncompliance or errors, drug interactions or side effects, use of over-the-counter medications such as nonsteroidal anti-inflammatory drugs (NSAIDs), substance abuse, uncontrolled diabetes or hypertension, infection, thyroid dysfunction, arrhythmias, myocardial ischemia, renal or hepatic insufficiency, pregnancy, and other physical or emotional stressors. At each visit, an assessment of the severity and triggers of dyspnea, fatigue, chest discomfort, palpitations, or presyncope should be performed.
Physical Examination
• Acute decompensated HF. The classic findings of acute decompensated “congestive” HF include a resting tachycardia, tachypnea, diffuse pulmonary rales, and an abnormal apical impulse (enlarged, diffuse, displaced, dyskinetic, or sustained).7 In acute, decompensated “low output” HF, particularly HF-rEF, systemic hypoperfusion may be manifest as by hypotension, a reduced pulse pressure or pulsus alternans, diminished carotid upstroke volume, Cheyne–Stokes respirations, cool extremities, and altered mentation. Whether this is a new onset diagnosis, or an acute decompensation of chronic HF, these patients will generally require acute hospitalization.
• Chronic HF. In chronic HF, it is very common to find fairly clear lung fields with coarse breath sounds or reduced respiratory diaphragmatic excursion.7 Bibasilar or diffuse rales are observed typically when filling pressures are rapidly or markedly elevated. Pleural effusions, when present, are more right-sided than left, or bilateral. The greater the number of symptoms and signs observed in a given patient, the more reliable is the diagnosis of HF. The most specific physical findings are an elevated jugular venous pressure, an S3, a laterally displaced apical impulse, pulmonary rales that do not clear with cough, and peripheral edema not due to primary venous insufficiency. Nonspecific physical findings include cardiomegaly or an abnormal apical impulse, an S4, and tachypnea.
• Signs of biventricular or predominant “right-heart” failure include an elevated jugular venous pressure, right ventricular (RV) parasternal lift or subxiphoid tap, RV gallop, loud P2 (pulmonary hypertension), abdominojugular reflux, pulsatile or tender hepatomegaly, ascites, and peripheral (dependent) edema. Signs of right-sided HF without signs of LV dysfunction may redirect your attention to primary or secondary pulmonary vascular diseases. Murmurs may reveal the cause of HF (valvular stenosis or regurgitation, hypertrophic cardiomyopathy with outflow tract obstruction) or in the case of mitral regurgitation, a possible consequence of LV remodeling and enlargement.7
Diagnostic Testing—Laboratory and Imaging
• Electrocardiography. The baseline electrocardiogram should be assessed for signs of prior infarction, ischemia, arrhythmia, conduction delays, and chamber enlargement or hypertrophy as these may provide clues to the underlying etiology of LV dysfunction. Low QRS voltage may indicate an occult primary or secondary infiltrative myocardial disease such as amyloidosis or a pericardial effusion. Nonspecific ST-T wave abnormalities are common. The QT/QTc interval may be prolonged, can reflect electrolyte abnormalities, myocardial disease, and drug effects, and confers an increased risk of ventricular arrhythmia.
• Chest radiography. It is important to note that a normal chest radiograph does not rule out the diagnosis of HF, but may afford a differential diagnosis. The chest x-ray can yield information on HF etiology and the degree of fluid overload or hemodynamic compensation. The cardiothoracic ratio and silhouette show that cardiac chambers are grossly enlarged. The amount of pulmonary vascular crowding, upper lobe redistribution, edema, Kerley B lines, or pleural effusions points more to volume status in the chronic setting and to the time course of hemodynamic alterations in the acute setting.
• Laboratory tests. The HF treatment guideline of the ACC/AHA recommends that all patients with HF initially undergo complete laboratory evaluation, including a complete blood count (CBC), serum electrolytes (including calcium, magnesium), blood urea nitrogen and serum creatinine, glucose, liver function tests, a fasting lipid profile, thyroid-stimulating hormone, and a urinalysis.1 Other laboratory tests such as HIV or other viral serologies, serum transferrin and iron saturation, and rheumatologic markers are obtained only if indicated by the history and physical examination. Serial measurements of electrolytes and renal function are typically advisable during medication titration.
• Serum biomarkers. For outpatients with complaints of dyspnea, measurement of brain natriuretic peptide (BNP) or N-terminal pro-BNP (NT-pro-BNP or BNPP) are well validated and useful tests to support the diagnosis of HF, as these peptides are synthesized and released by the heart primarily in response to hemodynamic perturbations.1 The level of BNP or BNPP correlates with disease severity and prognosis in both ambulatory outpatients and acute decompensated hospitalized patients, such that its measurement is a class 1 recommendation in HF treatment guidelines.1 However, natriuretic peptides have not been shown to be effective in screening and identifying asymptomatic patients with ventricular dysfunction. Elevations in plasma BNP levels are seen in acute, decompensated and chronic HF, acute MI, myocardial ischemia, and LV hypertrophy. The normal ranges for BNP and NT-proBNP are higher in women than in men, and in both sexes increase with age and/or declining renal function.8 In contrast, in the setting of morbid obesity, BNP levels may be disproportionately low. Marked elevations in BNP levels correlate with symptoms and the degree of LV systolic dysfunction (EF). However, a modestly elevated BNP level can occur in other settings, such as post-cardioversion, cardiac surgery, anemia, pulmonary embolism, pulmonary hypertension, renal failure, bacterial sepsis, severe burns, and other critical illnesses. Cardiac troponin levels can be elevated in decompensated HF patients without evidence of active myocardial ischemia or acute coronary syndromes, including patients without coronary artery disease (CAD). Measurement of this indicator of myocardial injury is recommended as part of the evaluation of patients with acute decompensated HF.1 Troponin elevations are usually mild but when present are associated with impaired hemodynamics, more severe LV dysfunction, worse clinical outcomes, and higher mortality rates.1,8–10 Newer, emerging biomarkers of myocardial fibrosis such as soluble ST-2 and galectin-3 are predictive of both hospitalization and death in HF and may be additive to natriuretic peptide levels. Multimarker assessment strategies as a means to guide treatment are being evaluated for relative efficacy in predicting change in prognosis over time in HF patients.8–10
• Echocardiography. The most valuable and cost-effective test in the diagnosis of HF is two-dimensional echocardiography with Doppler imaging, which facilitates the detection of abnormalities in myocardial, valvular, and pericardial structure and function.11 One major determinant of the appropriate course of therapy for HF is whether the LV EF is preserved or reduced. This information is quantified by echocardiography, along with cardiac chamber dimensions and/or volumes, LV wall thickness, and ventricular diastolic filling dynamics. Further, echocardiography provides an estimation of intracardiac hemodynamics, an evaluation of chamber geometry and assesses regional wall motion. The preference for echocardiography as an imaging modality is based upon its nearly ubiquitous availability and imaging quality without the use of ionizing radiation. Alternatively, the LV or RV EF and ventricular filling dynamics can also be determined by radionuclide imaging techniques. Cardiac magnetic resonance (CMR) imaging and cardiac computed tomography (CT) are also increasingly useful modalities in evaluating ventricular size, function and mass, detecting intracardiac shunts, RV dysplasia and other anatomical abnormalities. Given their cost, inherent radiation exposure, and imaging limitation at elevated heart rates, the routine use of these modalities has been limited. CMR and CT can be protocoled in order to distinguish viable myocardium from ischemic, infarcted, or fibrotic scar tissue. Ischemia and viability assessments by these techniques may be an important tool in determining whether to refer HF-rEF patients with known CAD for surgical revascularization.12
• Other diagnostic testing. Once the clinical diagnosis of HF is confirmed with supportive data from echocardiography, the remainder of diagnostic testing is directed at determining the underlying etiology. Irrespective of LV EF, in all patients with HF, the etiology that is most important to consider and exclude is CAD. Strategies involving noninvasive stress ischemia evaluation or coronary angiography are best chosen based on symptoms, signs, and CAD risk factors. With respect to acute decompensated HF, the use of invasive hemodynamic monitoring is recommended for those patients with poor perfusion and/or severe dyspnea in whom clinical assessment is unable to assess fluid status, hemodynamics, and cardiac output.1
Prognostic Assessment
An assessment of prognosis should be considered an integral part of the evaluation of a patient with HF. Risk assessment is recommended at the time of diagnosis and periodically thereafter. There are a number of well-validated multivariable risk scores available to help estimate an individual patient’s risk of mortality, both in the ambulatory and acute care settings, for both HF-rEF and HF-pEF populations, although their utilization is considered a class IIA guideline recommendation.1 In the absence of access to these score models or nomograms, each of the following is an easy-to-measure variable that lends independent, additive prognostic information.13
• Extent of LV dysfunction. LV EF less than 0.35 with lower values worse, significant LV enlargement, dilation, or concomitant restrictive filling dynamics (significant diastolic dysfunction) denote an extremely high-risk patient. Concomitant RV enlargement or dysfunction worsens prognosis further.
• Symptom class. Risk worsens with higher NYHA class, with NYHA IV having a 30% to 50% annual mortality risk. Persistent moderate to severe HF symptoms despite standard medical therapy warrants consideration of patient referral to an HF specialist.
• Hemodynamics. Clinical, echocardiographically estimated or measured pulmonary hypertension in the setting of LV systolic dysfunction carries a worse prognosis and is an indication for more aggressive therapy.
• Exercise capacity. Although age dependent, the inability to walk more than 300 m in a 6-minute walk test (for any reason) infers substantially greater annual risk of death or morbidity compared with a patient who can walk 450 m or more. Markedly impaired oxygen consumption with exercise, measured as a VO2 max <15 mL/kg/minute, or achieving less than 4 to 5 metabolic equivalents (METS) of work on bicycle or treadmill cardiopulmonary exercise test has a markedly adverse prognosis. Significant exercise impairment corresponds to a 1-year mortality rate of 20% or higher.
• Arrhythmia. Atrial fibrillation, atrial or ventricular tachyarrhythmias, or evidence of other conduction system disease such as arteriovenous (AV) nodal block or left bundle branch block worsen prognosis. Any family history of sudden death is associated with worse prognosis. Approximately 50% to 70% of patients with low EF and symptomatic HF have episodes of nonsustained ventricular tachycardia on routine ambulatory electrocardiographic monitoring; this is generally not indicated for screening purposes in the absence of symptoms.
• Hyponatremia. Serum sodium concentration of 135 mg per dL or less is generally related to intense renin–angiotensin system activation and denotes a higher-risk patient.
• Chronic kidney disease. Significant renal insufficiency not due to expected (reversible) medication effects is associated with worse outcomes in HF. Worsening renal function typically defined as an increase in serum creatinine ≥0.2 mg per dL or a corresponding decrease in estimated glomerular filtration rate ≥5 mL × min × 1.73 m2 is an adverse sign predicting substantially higher rates of mortality and hospitalization in patients with HF.
• Anemia. Anemia is present in up to 35% of HF patient populations. In studies that analyzed hemoglobin as a continuous variable, a 1-g per dL decrease in hemoglobin was independently associated with significantly increased mortality risk. Anemia can worsen cardiac ischemia, impair cardiac function and is associated with poor outcomes, including a higher risk of hospitalization, decreased exercise capacity, and poor quality of life.
Assessment of Comorbidities
Patients and practitioners often underestimate the substantial influence of comorbid diagnoses or conditions on the clinical course and stability of patients with HF. These common conditions include atherosclerosis, diabetes, hypertension, hyperlipidemia, thyroid dysfunction, anemia, obstructive sleep apnea, depression, and obesity. Concurrent infections can trigger HF decompensation due to fever and physiologic stressors and should be treated early and aggressively. Treating these conditions optimally may reduce ongoing or limit additional myocardial injury as well as reduce hospitalizations and improve outcomes. In particular, sleep-disordered breathing and depression are very common in HF populations and worsen clinical outcomes and quality of life. Immunizations and general health care maintenance should be kept up to date.
Differential Diagnosis
The differential diagnosis of a patient with prominent dyspnea ± edema includes pulmonary parenchymal disease (obstructive vs. interstitial), pulmonary thromboembolic disease, cor pulmonale, pulmonary veno-occlusive disease, primary or other secondary pulmonary arterial hypertension, exertional asthma, severe anemia, mitral stenosis, neuromuscular disease, constrictive pericarditis, or metabolic causes (i.e., acidosis). The differential diagnosis of a patient with predominant edema ± dyspnea includes severe venous insufficiency, nephrotic syndrome, cirrhosis, lymphedema, combined vascular insufficiency, and adverse medication effects (i.e., dihydropyridine calcium-channel blockers).
TREATMENT
Pharmacologic Management of Chronic HF-rEF
The majority of clinical research trials that have established the foundation of traditional medical therapy have focused on HF with systolic LV dysfunction (LVD). The implicit goals of treating chronic HF are to (a) improve patient symptoms and quality of life, (b) slow or reverse the progression of cardiac dysfunction, and (c) reduce HF mortality, morbidity, and therefore the cost burden of acute care. Since the pathophysiology of HF is complex, so follows the pharmacologic regimen. Angiotensin-converting enzyme inhibitors (ACE-I) and β-blockers have become the cornerstone of therapy to delay, halt, or reverse cardiac remodeling and improve mortality. In addition, the roles of diuretic therapy, aldosterone inhibition, digoxin, and other vasodilator therapy are reviewed.
Angiotensin-Converting Enzyme Inhibitors
Contemporary treatment guidelines for systolic LVD mandate that an ACE-I be utilized as primary therapy unless contraindicated.1,14 ACE-I improve hemodynamics by reducing afterload and attenuate the vasoconstrictor activity of angiotensin II (Ang II). Ang II also has thrombogenic, atherogenic, profibrotic, and other effects that contribute to progressive LV remodeling. ACE-I improve HF symptoms and quality of life. The progression of HF is slowed by ACE-I therapy, as evidenced in clinical trials by a survival benefit and fewer hospitalizations. ACE-I are indicated for use in the primary prevention of HF in patients at risk, post-MI patients regardless of EF, and in patients with documented LVD regardless of symptoms (AHA Stages A to D).1
ACE-I are typically initiated at low dose and uptitrated over days to weeks until side effects are noted or the dose reaches the equivalent of those used in the HF trials (Table 9.4-2). In general, higher achieved doses result in greater reductions in morbidity and hospitalization for HF, but mortality reduction is seen at virtually all doses. Electrolytes and renal function should be checked before initiation, after every dose increase, and after addition of other medications. Hypotension is seen most frequently during the first few days of initiation or dose increase, particularly in patients with hypovolemia, a recent large diuresis, or severe hyponatremia (serum sodium under 130 mmol per L). ACE-I lower blood pressure and alter intra-renal hemodynamics, inducing a predictable increase in serum creatinine. A modest elevation and plateau in blood urea nitrogen and serum creatinine concentration are expected with the use of diuretics and/or vasodilator therapy in HF. Progressively worsening renal function, however (i.e., serum creatinine increase of more than 0.3 mg per dL over a normal baseline, or a serum creatinine >2.5 to 3.0 mg per dL, may represent renal hypoperfusion due to a reduction in cardiac output or renal perfusion pressure.
In the absence of hyperkalemia, most HF experts will still initiate ACE-I therapy in a patient with a serum creatinine ≤3 mg per dL, employing an agent that is hepatically cleared to prevent drug or metabolite accumulation and profound hypotension. Other possible side effects of ACE-I include orthostatic hypotension, dizziness, hyponatremia, cough, and angioedema. Cough develops in 5% to 10% of patients on an ACE-I but is much more common, up to 50%, in Asian/Chinese populations. Cough represents the most common reason for drug withdrawal. It should be noted that cough might also represent worsening HF or other conditions, so if not resolved after a temporary discontinuation of the drug, the ACE-I should be restarted. Angioedema can be mild, or life-threatening in severe cases. Its prevalence is estimated at <1%, but is more common in black populations.
Angiotensin-Converting Enzyme Inhibitors and Angiotensin Receptor Blockers Commonly Used for the Treatment of Chronic HF (Generic Names Listed) |
| Starting dose | Maximum dose |
ACE-I |
|
|
Captopril | 6.25 mg 3 times daily | 50 mg 3 times daily |
Enalapril | 2.5 mg twice daily | 10–20 mg twice daily |
Fosinopril | 5–10 mg daily | 40 mg daily |
Lisinopril | 2.5–5 mg daily | 20–40 mg daily |
Perindopril | 2 mg daily | 8–16 mg daily |
Quinapril | 5 mg twice daily | 20 mg twice daily |
Ramipril | 1.25–2.5 mg daily | 10 mg daily |
Trandolapril | 1 mg daily | 4 mg daily |
ARBs |
|
|
Candesartan | 4–8 mg daily | 32 mg daily |
Irbesartan | 75 mg daily | 150–300 mg |
Eprosartan | 300 mg daily | 800 mg daily |
Losartan | 25–50 mg daily | 50–100 mg daily |
Olmesartan | 10 mg daily | 20–40 mg daily |
Telmesartan | 20 mg daily | 40–80 mg daily |
Valsartan | 20–40 mg twice daily | 160 mg twice daily |
Angiotensin Receptor Blockers
This class of drugs has become an acceptable alternative to ACE-I for both the prevention and treatment of HF.1 The most convincing data regarding the efficacy of angiotensin receptor blockers (ARBs) in HF therapy derives from the recent Candesartan in Heart Failure Assessment of Reduction in Mortality and Morbidity Trial (CHARM). The CHARM study had three placebo-controlled arms, one evaluating candesartan as an alternative to an ACE-I, the second as added therapy, and the third as therapy in patients with HF-pEF. In each arm, a therapeutic benefit from the ARB was observed.15,16
Like ACE-I, ARBs are also initiated at low dose and uptitrated over days to weeks (Table 9.4-1). An ARB is the best substitute for an ACE-I when the latter induces cough. The risk of angioedema is much lower with ARBs, but has been observed in approximately 8% of patients given an ARB after developing angioedema to the former drug. Other ARB side effects are quite similar to those of the ACE-I class of agents. Aside from the pathway of drug metabolism, little difference exists between the available ACE-I and ARBs, the choice of agent becomes clinician or formulary preference.
β-Blockers
The pivotal trials that provided incontrovertible evidence of the efficacy of β-blocker therapy in patients with chronic HF were the U.S. Carvedilol Trials Program, Cardiac Insufficiency Bisoprolol Study-2 (CIBIS-II), and the Metoprolol CR/XL Randomized Intervention Trial in Congestive Heart Failure (MERIT-HF) study.17–19 HF guidelines state that only carvedilol, sustained release metoprolol succinate, or bisoprolol should be added to background ACE-I or ARB therapy (Table 9.4-3). The reason for this is that β-blockers are a very heterogeneous group of agents, exhibiting variable selectivity for β-receptors, marked differences in pharmacokinetics, and many have ancillary vasodilating or additional antioxidant properties. First, the patient should be carefully evaluated for clinical stability and should be considered euvolemic.
β-Blockers should be started at low dose and in general, uptitrated approximately every 2 weeks, with target doses reached in about 8 to 12 weeks. Patients should be followed carefully for signs of impending decompensation or side effects (fluid retention, hypotension, dyspnea, fatigue, bradycardia, or heart block). Patients who manifest worsening HF symptoms or fluid retention should have diuretic medications adjusted. A temporary dose reduction in ACE-I, ARB, or other vasoactive medications may be necessary when symptomatic hypotension is present. With careful observation the vast majority of patients can tolerate β-Blockers therapy and achieve target doses. In general, patients on chronic β-Blockers therapy experiencing an acute decompensation should remain on their β-blocker, or be given a reduced dose and uptitrated again following symptomatic resolution. Avoid abrupt discontinuation, especially in patients with underlying ischemic heart disease. If the initiation or uptitration of β-blockers to target doses proves difficult, consider referral to an HF specialist.
β-Adrenergic Blocking Drugs Approved for the Treatment of Chronic HF-rEF |
β-Blocker | Initial dose and typical increment during uptitration | Treatment dose goal in clinical trials |
Bisoprolol | 1.25 mg daily | 10 mg daily |
Carvedilol | 3.125 mg twice daily | 25 mg twice daily 50 mg twice daily if >85 kg |
Metoprolol CR/XL | 12.5–25 mg daily | 200 mg daily |
Other Vasodilators
For patients with HF and LV systolic dysfunction intolerant of ACE-I or ARB therapy (due to renal dysfunction or hyperkalemia), the combination of hydralazine (H) and isosorbide dinitrate (ISDN) is recommended.1,20 The mortality benefit with H-ISDN is not as great as that seen with ACE-I or ARB but is significantly better than those of placebo or vasodilatory α-blockers (prazosin). The H-ISDN combination is appealing in chronic kidney disease, as it tends to increase renal cortical blood flow. Side effects including headache, dizziness, diarrhea, tachycardia, and somnolence are significant with this regimen; up to 25% of patients will discontinue one or both. The benefit of nitrates is presumed to be related to enhanced nitric oxide bioavailability. Hydralazine is a direct-acting vasodilator, but also reduces nitrate tolerance. Nitrate therapy is useful in decreasing orthopnea and PND and tends to improve exercise tolerance in patients who have persistent limitations despite optimization of other therapies.
The African-American Heart Failure Trial (A-HeFT) enrolled 1,050 self-identified African American patients who had NYHA class III or IV HF with LV dilation and systolic dysfunction.21 A new fixed-dose combination of H-ISDN (or placebo) was utilized in addition to background therapy of an ACE-I or ARB, a β-blocker, and a loop diuretic. Many patients were also taking digoxin and an aldosterone antagonist. Patients receiving the H-ISDN combination demonstrated a 43% reduction in all-cause death and a 33% reduction in first HF hospitalizations and a significant improvement in quality of life. The fixed-dose combination (BiDil) includes 37.5 mg H and 20 mg ISDN, and was titrated to 225/120 mg per day in three divided doses in this study.
The Prospective Randomized Amlodipine Survival Evaluation Trial-2 (PRAISE-2) added amlodipine or placebo to background HF therapy of an ACE-I, digoxin, and diuretics in nonischemic cardiomyopathy and symptomatic HF.22 There was no survival advantage (or disadvantage) attributable to amlodipine administration in this population, and very little data regarding patients also taking β-blockers. Calcium-channel blockers are not indicated as routine treatment for HF in patients with current or prior symptoms of HF and reduced LV EF, but amlodipine could be considered for the management of hypertension.1
Aldosterone Inhibitors
The Randomized Aldosterone Evaluation Study (RALES) demonstrated a beneficial effect on mortality due to progressive HF from low-dose spironolactone in patients with a recent hospitalization, and continued moderate to severe HF symptoms (NYHA class III or IV) on ACE-I or ARB therapy.23 The Eplerenone in Mild Patients Hospitalization and Survival Study in Heart Failure (EMPHASIS-HF) found a reduction in the rate of death from any cause or hospitalization for HF in patients with NYHA class II symptoms.24
Clinical guidelines recommend aldosterone receptor antagonists in normokalemic patients with serum creatinine less than 2.5 mg per dL with NYHA class II to IV HF and LV EF less than 0.35. Aldosterone receptor antagonists are also recommended to reduce morbidity and mortality following an acute MI in patients with LV EF of less than 0.40 who develop symptoms of HF based on findings in the EPHESUS trial.1,23 Serum sodium and potassium should be checked at 1 week, frequently thereafter, including any time there is a change in dosage of any medication that may influence potassium balance or renal function. Strong consideration should be given to lowering or eliminating supplemental potassium when spironolactone is added to the regimen, to lessen the risk of potentially fatal hyperkalemia.
Digitalis
Digoxin remains a controversial drug in HF with systolic LVD two centuries after its initial use. Digoxin withdrawal from patients with stable chronic HF on an ACE-I contributes to decompensation requiring treatment or hospital admission, reduced exercise capacity, and lower quality-of-life scores.25 In the Digitalis Investigation Group (DIG) trial, the addition of digoxin to baseline therapy had a neutral effect on mortality but decreased hospitalizations for HF. Digoxin increases EF by 3% to 5% due to its positive inotropic effects mediated by sodium–potassium pump inhibition.26 More importantly, digoxin improves rest and exercise LV hemodynamics, and is sympathoinhibitory, attenuating the neurohormonal and baroreceptor abnormalities seen in chronic HF. This latter role as neurohormonal antagonist may be the more important mechanism of action, as all other agents with positive inotropic activity have increased HF mortality in clinical trials.
Digoxin therapy may be limited by renal insufficiency and conduction abnormalities (heart block, slow atrial fibrillation) but is generally well tolerated, with few side effects at recommended doses. Current guidelines state its use should be considered if HF symptoms remain after ACE-I and β-blockers titration when the EF remains less than 0.40 or for patients with severe symptoms who have not yet responded symptomatically to optimal medical therapy.1 Loading doses are unnecessary, and most patients should be prescribed a dose of 0.125 mg daily. Current guidelines suggest doses of digoxin that achieve a plasma concentration of drug in the range of 0.5 to 0.9 ng per mL. There have been no prospective, randomized studies of the relative efficacy or safety of different plasma concentrations of digoxin. Overt digoxin toxicity is commonly associated with serum digoxin levels greater than 2 ng per mL but can occur at lower levels, especially in the setting of hypokalemia, hypomagnesemia, or hypothyroidism.26,27 Digoxin trough levels should be obtained if there is clinical suspicion of toxicity due to signs or symptoms and to determine whether dose (or dose frequency) reduction is indicated in the setting of worsening renal function or loss of lean body mass. The initiation of medications with known or possible interactions that may increase digoxin concentrations (verapamil, quinidine, amiodarone, spironolactone, and certain antibiotics) should be coupled with measurement of digoxin level within 1 week of initiation of the drug to ensure the digoxin level does not exceed 1 ng per mL.1
Commonly Used Diuretic Drugs for the Treatment of Sodium and Fluid Retention in Chronic HF |
| Initial oral dose | Recommended maximal oral dose |
Thiazide class |
|
|
Chlorothiazide | 250 mg daily | 500 mg daily |
Hydrochlorothiazide | 25 mg daily | 50–100 mg daily |
Metolazone | 2.5 mg daily | 10 mg daily |
Loop diuretics |
|
|
Furosemide | 10–40 mg daily or twice daily | 200 mg daily or twice daily |
Bumetanide | 0.5–1.0 daily or twice daily | 4–5 mg daily or twice daily |
Torsemide | 10 mg daily | 200 mg daily |
Diuretics
Diuretic therapy is indicated for symptoms or signs of systemic or pulmonary congestion due to volume overload.28–30 Diuretics relieve congestive symptoms by promoting excretion of excess sodium and therefore water (Table 9.4-4). There are no controlled clinical trial data prospectively evaluating the overall impact of diuretic therapy on mortality in patients with HF. Diuretics promote activation of the RAAS, potentiate the hypotensive effects of ACE-Is/ARBs, and may decrease cardiac output, especially in patients with diastolic LVD. Chronic diuretic therapy can be limited by the development of diuretic resistance or refractoriness. Diuretics also induce hypokalemia, hypomagnesemia, and hyperuricemia, and promote calciuria. Electrolytes require close monitoring when diuretics are used.30
Thiazide diuretics should be used if fluid retention is mild, but are effective only when the glomerular filtration rate is greater than 30 mL per minute. Loop diuretics are the mainstay of diuretic therapy in HF when congestion is moderate. When fluid retention is extreme or the patient has become refractory to loop diuretics, intravenous administration of a loop diuretic or the addition of the thiazide-like agent metolazone (1 to 5 mg 30 minutes to 1 hour prior to loop agent) can dramatically increase natriuresis and induce severe electrolyte loss. Potassium-sparing diuretics should be used with caution in patients on ACE-Is or ARBs and in patients with diabetes prone to type IV renal tubular acidosis.
Pharmacologic Management of Chronic HF-pEF
Trials using comparable and efficacious agents for HF-rEF have generally been disappointing when used in patients with HF-pEF.1,31–33 Therefore, most of the recommended therapies for HF with diastolic LVD are directed at symptoms, especially comorbidities, and risk factors that may worsen cardiovascular disease. CAD and hypertension should be aggressively treated, if present. Blood pressure control remains the most important recommendation in patients with HF-pEF as it has been shown to reduce hospitalizations for HF.34 β-Blocking agents, ACE and ARBs are all reasonable to employ for this purpose.1
Diuretics are typically necessary for congestive symptoms, but excessive preload reduction (nitrates, diuretics) can impair cardiac output and exacerbate hypotension and should be used with caution. There is no recommended role for digoxin therapy in a patient with HF-pEF. Patients with atrial fibrillation and rapid ventricular response intolerant of or refractory to β-blocker or amiodarone therapy should be referred to an electrophysiologist for possible catheter ablation.
Nonpharmacologic HF Therapies
Patient Education/Behavioral Interventions
Discuss with the patient and family the diagnosis and reason(s) for the development of HF, including estimated prognosis and intended treatment plan. Symptoms referable to HF should be reviewed and patients instructed to call if symptoms are noted or increased, particularly rapid weight gain or loss. Emphasize sodium restriction to 1,500 mg per day, along with daily weight monitoring. Stress the importance of medication adherence, good nutrition, and physical activity. Fluid restriction is generally necessary only when excessive or when volume status is difficult to manage with diuretics and sodium restriction. Fluid restriction is advisable in the setting of severe hyponatremia, however. Smoking cessation should be advocated.1
Guidelines recommend that patients’ literacy, cognitive status, psychological state, culture, and access to social and financial resources be taken into account for optimal education and counseling.1 It is also recommended that providers frequently assess for medication nonadherence or dietary noncompliance (sodium, alcohol, excess fluids), drug abuse, and medication additions by other providers (calcium-channel blockers in HF-rEF, NSAIDs, glitazones, remicade, and certain over-the-counter medications or supplement use35). It is important to confirm and treat uncontrolled hypertension, diabetes, paroxysmal arrhythmias, depression, or sleep apnea. Surveillance laboratory testing is helpful in detecting decreasing renal or hepatic function, new or worsening anemia, and occult infection.
Written educational materials are quite useful and downloadable from the Heart Failure Society of America Web site (www.hfsa.org or www.abouthf.org) and the American Heart Association Get with the Guidelines program. Consider referral to a dietician or disease management program when patient understanding is an impairment to the success of your plan of care. Such disease management programs have been shown to significantly reduce HF hospitalizations, produce shorter lengths of stay, lower health care costs, and improve both quality of life and survival.36
Exercise and Cardiac Rehabilitation
Several controlled trials have shown that exercise training can reduce symptoms, increase exercise capacity, and improve the quality of life of patients with chronic HF. Exercise prescriptions (aerobic and light resistance training) are generally safe in compensated HF. In the Heart Failure: A Controlled Trial Investigating Outcomes of Exercise Training (HF-ACTION) trial, 2,331 medically stable outpatients with HF with reduced EF were randomized to exercise training for 3 months or to standard medical therapy. When adjusted for CAD risk factors, patients assigned to the exercise group had a significant reduction in all-cause mortality, cardiovascular mortality, and hospitalizations.37 On the basis of clinical guidelines, cardiac rehabilitation is recommended in clinically stable patients with HF in order to improve functional capacity, exercise duration, health-related quality of life, and mortality.1
Treatment of Sleep Disorders
Sleep disorders are common in patients with HF. A study of adults with chronic HF treated with evidence-based therapies found that 61% had either central or obstructive sleep apnea.38
The primary treatment of obstructive sleep apnea (OSA) is nocturnal continuous positive airway pressure (CPAP). In a major trial, CPAP for OSA was effective in decreasing the apnea–hypopnea index, improving nocturnal oxygenation, increasing LV EF, lowering norepinephrine levels, and increasing the distance walked in 6 minutes. These benefits were sustained for up to 2 years.39 Clinical guidelines therefore recommend maintaining a high index of suspicion for sleep disorders in HF patients and referring patients for a sleep study when indicated.1
Biventricular Pacemakers
Dyssynchronous contractions between the LV and RV can be improved by electrically activating the right and left ventricles in a sequential manner with a biventricular pacemaker device. Cardiac resynchronization therapy (CRT) appears to improve EF, reduce secondary mitral regurgitation, and improve HF symptoms (when moderate to severe), as well as enhance exercise capacity and quality of life.40–42 There is strong evidence to support the use of CRT to improve survival and to decrease hospitalizations in patients with persistently symptomatic HF receiving optimal medical therapy who have cardiac dyssynchrony (evidenced by a prolonged QRS duration greater than 120 ms) and an EF of 0.35 or less. Based on the Multicenter Automatic Defibrillator Implantation Trial-Cardiac Resynchronization Therapy (MADIT-CRT) trial, CRT may also be considered for patients with class I symptoms greater than 40 days after MI with EF less than 0.30, sinus rhythm, left bundle branch block, and QRS duration greater than 150 ms.43 CRT should not be considered as “rescue” therapy for Stage D HF.1
Automated Implanted Cardiac Defibrillators
Patients with HF-rEF are at increased risk for ventricular tachyarrhythmias leading to sudden cardiac death. Current guidelines recommend prophylactic implantation of automated implanted cardiac defibrillators (AICD) in patients with nonischemic cardiomyopathy or ischemic heart disease with an EF less than 35% and mild to moderate HF symptoms (NYHA II or III) when at least 1-year survival with good functional capacity is expected.1 AICD implantation is also recommended in patients at least 40 days post-MI with EF less than 0.30 and NYHA class I symptoms.40,43 Again, optimal medical therapy should be previously employed and demonstrate a persistent reduction in EF. AICDs are generally not warranted in patients with refractory HF (Stage D) or in patients with concomitant diseases that shorten life expectancy independent of HF. AICDs have bradycardia and anti-tachycardia pacing capabilities as well. Although highly effective in preventing sudden death, frequent shocks (appropriate or inappropriate) reduce quality of life and increase patient anxiety. Of note, antimicrobial prophylaxis is not recommended before dental, gastrointestinal, or genitourinary procedures to prevent device infection.44
Mechanical Circulatory Support Devices and Cardiac Transplantation
Cardiac transplantation is considered the gold standard for the treatment of refractory (Stage D) HF. Since the first cardiac transplantation in 1967, advances in immunosuppressive therapy have greatly improved the long-term survival of transplant recipients as well as their functional status and health-related quality of life.45 Selected patients with Stage D HF and poor prognosis should be referred to an advanced HF treatment program or cardiac transplantation center for evaluation.46
Mechanical circulatory support (MCS) devices have emerged as a viable therapeutic option for patients with advanced Stage D HF with reduced EF refractory to optimal medical therapy. MCS can be used as (a) bridge to transplantation and a decision regarding transplantation candidacy and for (b) “destination” or permanent therapy. Bridge to transplantation and destination therapy have the strongest database with regard to survival, functional capacity, and health-related quality of life benefits. Use of nondurable or temporary MCS (i.e., percutaneous or extracorporeal ventricular assist devices) is reasonable as a “bridge to recovery” or “bridge to decision” for carefully selected unstable patients with low EF with acute, profound hemodynamic compromise.47,48
Advanced Directives and End-of-Life Care
It is mandatory that discussions about advance directives occur in context with prognosis. These conversations are best performed in the office setting following the initial HF diagnosis, and after hospitalizations or change in clinical status. Including the spouse or a close family member is preferable. Use of prognosis modeling algorithms previously discussed is often helpful.49 Referral to an Advanced Heart Failure Treatment Program or Palliative Care Program for a confirmatory opinion is considered. Palliative care focuses on relief of pain and discomfort, provides emotional support for patient and family, and assists with transitions of care when quality of life can no longer be maintained. Patients with advanced (Stage D) HF can be enrolled in palliative care yet receive life-prolonging therapies such as inotropic drug infusions or destination therapy MCS.50 Hospice care focuses on symptom management while discontinuing life-prolonging medicine or treatment that does not improve patients’ quality of life. Some patients may choose deactivation of their AICD to avoid the discomfort of frequent shocks. Utilization of end-of-life care services such as hospice should occur after full and appropriate application of evidence-based pharmacologic and nonpharmacological treatments.51
REFERENCES
1. Yancy CW, Jessup M, Bozkurt B, et al. 2013 ACCF/AHA guideline for the management of heart failure: a report of the American College of Cardiology Foundation/American Heart Association Task Force on Practice Guidelines. J Am Coll Cardiol 2013;62(16):e147–e239.
2. Krumholz HM, Merrill AR, Schone EM, et al. Patterns of hospital performance in acute myocardial infarction and heart failure 30-day mortality and readmission. Circ Cardiovasc Qual Outcomes 2009;2:407–413.
3. Go AS, Mozaffarian D, Roger VL, et al. Heart disease and stroke statistics–2013 update: a report from the American Heart Association. Circulation 2013;127:e6–e245.
4. Curtis LH, Whellan DJ, Hammill BG, et al. Incidence and prevalence of heart failure in elderly persons, 1994–2003. Arch Intern Med 2008;168:418–424.
5. Cohn JN, Ferrari R, Sharpe N. Cardiac remodeling-concepts and clinical implications: a consensus paper from an international forum on cardiac remodeling. J Am Coll Cardiol 2000;35:569–582.
6. Greenberg B, Hermann D. Contemporary diagnosis and management of heart failure. 3rd ed. Newtown, PA: Handbooks in Healthcare; 2005.
7. Chatterjee K. Physical examination in heart failure. In: Hosenpud JD, Greenberg BH, eds. Congestive heart failure, pathophysiology, diagnosis and comprehensive approach to management. 3rd ed. Philadelphia, PA: Lippincott Williams & Wilkins; 2007:615–627.
8. Maisel AS, Bhalla V, Braunwald E. Cardiac biomarkers: a contemporary status report. Nat Clin Pract Cardiovasc Med 2006;3(1):24–34.
9. Horwich TB, Patel J, MacLellan WR, et al. Cardiac troponin I is associated with impaired hemodynamics, progressive left ventricular dysfunction, and increased mortality rates in advanced heart failure. Circulation 2003;108:833–838.
10. Januzzi JL Jr. Use of biomarkers to “guide” care in chronic heart failure: what have we learned (so far)? J Card Fail 2011;17:622–625.
11. Vitarelli A, Tiukinhoy S, Di LS, et al. The role of echocardiography in the diagnosis and management of heart failure. Heart Fail Rev 2003;8:181–189.
12. Fihn SD, Gardin JM, Abrams J, et al. 2012 ACCF/AHA/ACP/AATS/PCNA/SCAI/STS guideline for the diagnosis and management of patients with stable ischemic heart disease: a report of the American College of Cardiology Foundation/American Heart Association Task Force on Practice Guidelines, and the American College of Physicians, American Association for Thoracic Surgery, Preventive Cardiovascular Nurses Association, Society for Cardiovascular Angiography and Interventions, and Society of Thoracic Surgeons. Circulation. 2012;126:e354–e471.
13. Hermann DD, Greenberg BH. Prognostic factors In: Poole-Wilson P, Colucci W, Massie B, et al, eds. Heart failure: scientific principles & clinical practice. New York, NY: Churchill Livingstone; 1997:439–454.
14. Garg R, Yusuf S. Overview of randomized trials of angiotensin-converting enzyme inhibitors on mortality and morbidity in patients with heart failure. Collaborative Group on ACE Inhibitor Trials. JAMA 1995;273(18):1450–1466.
15. Granger CB, McMurray JJ, Yusuf S, et al. Effects of candesartan in patients with chronic heart failure and reduced left-ventricular systolic function intolerant to angiotensin-converting-enzyme inhibitors: the CHARM-Alternative Trial. Lancet 2003;362:772–776.
16. McMurray JJ, Ostergren J, Swedberg K, et al. Effects of Candesartan in patients with chronic heart failure and reduced left-ventricular systolic function taking angiotensin-converting-enzyme inhibitors: the CHARM-Added Trial. Lancet 2003;362:767–771.
17. MERIT-HF Study Group. Effect of metoprolol CR/XL in chronic heart failure: metoprolol CR/XL randomized intervention trial in congestive heart failure. Lancet 1999;353:2001–2007.
18. Packer M, Bristow MR, Cohn JN, et al. The effect of carvedilol on morbidity and mortality in patients with chronic heart failure. N Engl J Med 1996;334:1349–1355.
19. CIBIS Investigators and Committees. The Cardiac Insufficiency Bisoprolol Study II (CIBIS II): a randomized trial of beta-blockade in heart failure. Lancet 1999;353:9.
20. Cohn JN, Johnson G, Ziesche S, et al. A comparison of enalapril with hydralazine-isosorbide dinitrate in the treatment of chronic congestive heart failure. N Engl J Med 1991;325:303–310.
21. Taylor AL, Ziesche S, Yancy C, et al. Combination of isosorbide dinitrate and hydralazine in blacks with heart failure. N Engl J Med 2004;351:2049–2057.
22. Packer M, O’Connor CM, Ghali JK, et al. Effect of amlodipine on morbidity and mortality in severe chronic heart failure. New Engl J Med 1996;335:1107–1114.
23. Packer M, Gheorghiade M, Young JB, et al. Withdrawal of digoxin from patients with chronic heart failure treated with angiotensin-converting enzyme inhibitors. RADIANCE Study. N Engl J Med 1993;329:1.
24. The Digitalis Investigation Group. The effect of digoxin on mortality and morbidity in patients with heart failure. N Engl J Med 1997;336:525.
25. Pitt B, Zannad F, Remme WJ, et al; Randomized Aldactone Evaluation Study Investigators. The effect of spironolactone on morbidity and mortality in patients with severe heart failure. N Engl J Med 1999;341:709–717.
26. Zannad F, McMurray JJ, Krum H, et al; for the EMPHASIS-HF Study Group. Eplerenone in patients with systolic heart failure and mild symptoms. N Engl J Med 2011;364:11–21.
27. Rathore SS, Curtis JP, Wang Y, et al. Association of serum digoxin concentration and outcomes in patients with heart failure. JAMA 2003;289:871–8.
28. Adams KF Jr, Patterson JH, Gattis WA, et al. Relationship of serum digoxin concentration to mortality and morbidity in women in the Digitalis Investigation Group trial: a retrospective analysis. J Am Coll Cardiol 2005;46:497–504.
29. Brater DC. Diuretic therapy. N Engl J Med 1998;339:387–395.
30. Dormans TJ, Gerlad PG, Russell FM, et al. Combination diuretic therapy in severe congestive heart failure. Drugs 1998;55(2):165–172.
31. Leier CV, Cas LD, Metra M. Clinical relevance and management of the major electrolyte abnormalities in congestive heart failure: hyponatremia, hypokalemia and hypomagnesemia. Am Heart J 1994;128:564–574.
32. Philbin EF, Rocco TA Jr. Use of angiotensin-converting enzyme inhibitors in heart failure with preserved left ventricular systolic function. Am Heart J 1997;134(2 pt 1):188–195.
33. Yusuf S, Pfeffer MA, Swedberg K, et al. Effects of candesartan in patients with chronic heart failure and preserved left-ventricular ejection fraction: the CHARM-Preserved Trial. Lancet 2003;362:777–781.
34. Edelmann F, Wachter R, Schmidt AG, et al. Effect of spironolactone on diastolic function and exercise capacity in patients with heart failure with preserved ejection fraction: the Aldo-DHF randomized controlled trial. JAMA 2013;309:781–791.
35. Piller LB, Baraniuk S, Simpson LM, et al. Long-term follow-up of participants with heart failure in the antihypertensive and lipid-lowering treatment to prevent heart attack trial (ALLHAT). Circulation 2011;124:1811–1818.
36. Hermann DD. Naturoceutical agents and cardiovascular medicine—the hope, hype and the harm. ACC Curr J Rev 1999;8(5):53–57.
37. Fonarow GC, Stevenson LW, Walden JA, et al. Impact of a comprehensive heart failure management program on hospital readmissions and functional status of patients with advanced heart failure. J Am Coll Cardiol 1997;30:725–732.
38. O’Connor CM, Whellan DJ, Lee KL, et al. Efficacy and safety of exercise training in patients with chronic heart failure: HF-ACTION randomized controlled trial. JAMA 2009;301:1439.
39. MacDonald M, Fang J, Pittman SD, et al. The current prevalence of sleep disordered breathing in congestive heart failure patients treated with beta-blockers. J Clin Sleep Med 2008;4:38–42.
40. Bradley TD, Logan AG, Kimoff RJ, et al. Continuous positive airway pressure for central sleep apnea and heart failure. N Engl J Med 2005;353:2025–2033.
41. Bristow MR, Saxon LA, Boehmer J, et al. Cardiac-resynchronization therapy with or without an implantable defibrillator in advanced chronic heart failure. N Engl J Med 2004;350:2140–2150.
42. Cleland JG, Daubert JC, Erdmann E, et al. The effect of cardiac resynchronization on morbidity and mortality in heart failure. N Engl J Med 2005;352:1539–1549.
43. Moss AJ, Hall WJ, Cannom DS, et al. Cardiac-resynchronization therapy for the prevention of heart-failure events. N Engl J Med 2009;361:1329–1338.
44. Zareba W, Klein H, Cygankiewicz I, et al; MADIT-CRT Investigators. Effectiveness of Cardiac resynchronization Therapy by QRS Morphology in the Multicenter Automatic Defibrillator Implantation Trial–Cardiac Resynchronization Therapy (MADIT-CRT). Circulation 2011;123:1159–1166.
45. Baddour LM, Epstein AE, Erickson CC, et al. Update on cardiovascular implantable electronic device infections and their management: a scientific statement from the American Heart Association. Circulation 2010;121:458–477.
46. Butler J, Khadim G, Paul KM, et al. Selection of patients for heart transplantation in the current era of heart failure therapy. J Am Coll Cardiol 2004;43:787–793.
47. Mehra MR, Kobashigawa J, Starling R, et al. Listing criteria for heart transplantation: International Society for Heart and Lung Transplantation guidelines for the care of cardiac transplant candidates2006. J Heart Lung Transplant 2006;25:1024–42.
48. Feldman D, Pamboukian SV, Teuteberg JJ, et al. The 2013 International Society for Heart and Lung Transplantation Guidelines for mechanical circulatory support: executive summary. J Heart Lung Transplant 2013;32:157–187.
49. Slaughter MS, Rogers JG, Milano CA, et al. Advanced heart failure treated with continuous-flow left ventricular assist device. N Engl J Med 2009;361:2241–2251.
50. Adler ED, Goldfinger JZ, Kalman J, et al. Palliative care in the treatment of advanced heart failure. Circulation 2009;120:2597–2606.
51. Goda A, Williams P, Mancini D, et al. Selecting patients for heart transplantation: comparison of the Heart Failure Survival Score (HFSS) and the Seattle heart failure model (SHFM). J Heart Lung Transplant 2011;30:1236–1243.
| Atrial Fibrillation and Other Supraventricular Tachycardias |
ATRIAL FIBRILLATION
General Principles
Epidemiology
Atrial fibrillation (AF) is one of the most common cardiac dysrhythmias. There are about 2.5 million patients with this condition in the United States. The prevalence of AF is strikingly related to age, affecting as many as 10% of those older than 75 years. As the median age of the U.S. population continues to increase, so does the prevalence of AF. AF is a considerable health burden in that it increases total mortality twofold, heart failure threefold, and stroke rates fivefold. It is responsible for 10% to 15% of all strokes in the United States.1–3
Classification
The following classification has been proposed by the 2001 American College of Cardiology/American Heart Association/European Society of Cardiology Board Task Force. Management of AF is dependent on recognition of the appropriate classification:
• Paroxysmal AF is self-terminating and lasts less than 7 days and usually less than 48 hours. It can further be subdivided into first-episode paroxysmal AF and recurrent paroxysmal AF. Therapy here should focus on prevention of recurrence.
• Persistent AF is not self-terminating and lasts longer than 7 days. This can again be first episode or recurrent. Therapy should focus on modulation of heart rhythm or rate and preventing recurrence.
• Permanent or chronic AF has been present for more than 1 year, and cardioversion either has not been attempted or has failed. Control of ventricular rate is the usually preferred therapeutic option.
Over a 5-year period, about 25% of patients with paroxysmal AF will progress to persistent AF; the likelihood is increased in patients with other risk factors that are discussed below.1–3
Etiology
Besides age, other independent risk factors for AF include valvular heart disease, heart failure, coronary heart disease, obesity, obstructive sleep apnea, hypertension, and diabetes. Some other potentially reversible causes of AF include any cause of arterial hypoxemia; hypokalemia; hypomagnesemia; acute alcohol consumption; pericarditis; myocardial infarction (MI); and hyperadrenergic states such as postoperative period, including postcardiac surgery, theophylline, or other stimulant toxicity and endocrinopathies (hypo/hyperthyroidism, pheochromocytoma). About 10% of patients with AF have none of the above identifiable risk factors; they are considered to have “lone” AF, which necessitates further classification according to the system listed above.1–4
Pathophysiology
The pathogenic property common to all risk factors for AF is diastolic dysfunction of the left ventricle, which in turn leads to left atrial dilatation, stretch, and fibrosis and subsequent vulnerability to AF.1
Diagnosis
Clinical Presentation
Symptoms can often be vague and include fatigue, lightheadedness, palpitations, breathlessness, and exercise intolerance. Younger and more active patients and those with paroxysmal AF are more likely to report symptoms. Acute presentations may include decompensated congestive heart failure and angina.
History and Physical Examination
Initial evaluation of the patient with AF includes a careful history and physical examination to assess for presence of symptoms, other comorbidities, and potentially reversible causes as noted above. An irregularly irregular pulse that is usually greater than 100 beats per minute (bpm) is characteristic of AF. “Slow” AF may indicate associated disease of the conduction pathway. It is important to assess if the patient is hemodynamically stable or unstable as this would dictate further course of management.
Laboratory Studies
• Basic laboratory workup, including screening thyroid tests, complete blood count, and comprehensive metabolic panel, are warranted, especially for a first episode of AF. Urine drug screening and medication levels in the blood may also be considered.
• All patients with AF should have an electrocardiogram (ECG) and a transthoracic echocardiogram (TTE) to help identify AF and quantify any underlying cardiovascular disease and guide subsequent management.
• The ECG demonstrates chaotic electrical activity with an irregularly irregular rate and rhythm. This is evidenced by constantly changing R–R intervals and no discernible P waves before the QRS complexes. The fibrillation waves are best seen in leads II, III, aVF, and V1. The fibrillation pattern may be fine or coarse. A few patients have fine AF with little evidence on the ECG. The irregular ventricular pattern should allow determination of AF. Some patients have coarse fibrillation waves, making it difficult to distinguish from atrial flutter. This can again be distinguished by the erratic ventricular response. The QRS complexes are narrow unless there is aberrant ventricular conduction. The ECG may also reveal evidence of acute myocardial ischemia, pre-excitation, sinus node or conduction system disease, or QT prolongation (Figure 9.5-1).1
• The TTE determines left atrial size (a predictor for AF recurrence), presence of any valvular and/or pericardial heart disease, and can also detect left ventricular hypertrophy and ventricular dysfunction that will influence management decisions. Transesophageal echocardiogram (TEE) is more sensitive for identifying atrial thrombi.
• Intracardiac electrophysiologic studies may be useful in young patients with idiopathic lone AF to detect underlying pre-excitation or conduction pathway disease.
• Stress testing is indicated only if the initial evaluation suggests the presence of ischemic heart disease.
Treatment
Immediate management of AF depends on the hemodynamic status of the patient. If unstable, immediate synchronized cardioversion is indicated. If stable, management decisions that need to be made when managing AF include:
• Whether to attempt to restore and then maintain sinus rhythm, that is, adopt a rhythm-control strategy, or
Figure 9.5-1. Atrial fibrillation: Note the characteristic irregularity in the R–R intervals and the absence of well-defined P waves preceding the QRS complexes.
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


