Malformation
Incidence per million live births
Ventricular septal defect (VSD)
4,482
Atrial septal defect (ASD)
1,059
Pulmonary stenosis
836
Patent ductus arteriosus (PDA)
782
Tetralogy of Fallot
577
Coarctation of the aorta
492
Atrioventricular septal defect (AV canal)
396
Aortic stenosis
388
Transposition of the great arteries (TGA)
388
Hypoplastic left heart syndrome (HLHS)
279
Truncus arteriosus (TA)
136
Total anomalous pulmonary venous connection
120
Bicuspid aortic valve
13,817
Table 27.2.
Congenital Malformations Organized by Type of Physiology
Left-to-right shunts | Right-to-left shunts | Obstructive |
|---|---|---|
ASD | TOF | Aortic coarctation |
VSD | Truncus arteriosus | Aortic stenosis/atresia |
PDA | Tricuspid atresia | Pulmonary stenosis/atresia |
♦
No identifiable cause in most cases
♦
Between 8 and 18% are associated with chromosomal abnormality; over 50 genes have been associated with congenital heart defects
♦
First trimester rubella infection can lead to patent ductus arteriosus and pulmonary stenosis
♦
Down syndrome associated with septal defects (atrial and ventricular), defects of atrioventricular valves
♦
Turner syndrome associated with coarctation of aorta
♦
Drugs, such as alcohol and thalidomide, can also lead to congenital heart abnormalities
♦
Clinical presentation include asymptomatic murmur, cyanosis, failure to thrive, heart failure, and shock
♦
Shunt lesions :
Left-to-right shunts
•
Portion of oxygenated blood from the lungs is shunted back to the lungs
•
Most important complications are pulmonary hypertension due to increased pulmonary blood flow and eventual right ventricular hypertrophy
•
Increased pulmonary vascular resistance leads to reversal of shunt with cyanosis (Eisenmenger syndrome)
Right-to-left shunts
•
Portion of deoxygenated blood from systemic veins return to the systemic arterial circulation bypassing the lungs
♦
Obstructive lesions :
Obstructions can occur at the level of the valves, ventricular outflow tracts, or great arteries
Aortic Stenosis
♦
Obstructive disease
♦
Approximately 6% of congenital heart abnormalities
♦
May assume valvular, subvalvular, and supravalvular forms
♦
Most common cause is bicuspid aortic valve, with typical clinical presentation of valve failure in early middle age
♦
In the neonate, more commonly caused by unicuspid/unicommissural aortic valve; may result in the development of endocardial fibroelastosis and manifest as heart failure in infancy
♦
Williams–Beuren syndrome produces arterial wall thickening which effectively produces supravalvular stenosis of the aortic root and ascending aorta
♦
Subvalvular forms due to subaortic membrane
Atrial Septal Defect (ASD)
♦
Produces a left-to-right shunt
♦
Comprises approximately 10% of congenital heart abnormalities
♦
Defects at the fossa ovalis are called ostium secundum (OS) defects
♦
Defects inferior to the fossa ovalis are called ostium primum (OP) defects, usually a component of atrioventricular canal defects and associated with mitral valve defects
♦
OS defect is much more common than OP defect
♦
OS defects often not detected in childhood because of lack of symptoms but may later present in adulthood
Coarctation of the Aorta
♦
Obstructive disease
♦
Approximately 7% of congenital heart abnormalities
♦
More common in males
♦
Coarctation located usually as a discrete lesion proximal, distal, or opposite the orifice of the ductus arteriosus
♦
May be a long segment or tubular narrowing of the aortic arch
♦
Patients develop systemic hypertension in the upper body and collateral flow to the distal aorta
♦
Frequent (12%) in Turner syndrome
♦
50% of cases associated with bicuspid aortic valve
♦
Approximately 60% will die (frequently of aortic rupture and dissection) by age 40 if not corrected
Hypoplastic Left Heart Syndrome
♦
Obstructive disease
♦
Spectrum of cardiac anomalies with small left atrium, mitral valve atresia or stenosis, aortic atresia, underdeveloped left ventricle, and ascending aorta
♦
Right atrium, right ventricle, and pulmonary artery often dilated
♦
Interatrial communication through a patent foramen ovale (PFO) or ASD often present
♦
Aortic atresia with normal-sized left ventricle is associated with the presence of a large ventricular septal defect (VSD)
♦
Male predominance
♦
C yanosis from birth, with high mortality within the first month of life
Patent Ductus Arteriosus (PDA)
♦
Left-to-right shunt
♦
Manifestations depend on the size of communication between aorta and pulmonary artery
♦
Small PDA results in a small left-to-right shunt with “machinery” murmur and mild symptoms
♦
Large PDA results in a large shunt leading to pulmonary hypertension, eventually evolving into a shunt reversal with cyanosis (Eisenmenger physiology )
Pulmonary Stenosis and Atresia
♦
Obstructive disease
♦
Approximately 7% of congenital heart abnormalities
♦
Caused usually by a dome-shaped valve with central perforation or dysplastic pulmonary valve with thick myxoid cusps
♦
Leads to right ventricular hypertrophy and systemic venous congestion
♦
In case of pulmonary atresia, the valve is replaced by a fibrous membrane, and the right ventricle will be hypoplastic; blood to the lungs is delivered through a PDA, rarely through multiple aortopulmonary collaterals
Tetralogy of Fallot
♦
Right-to-left shunt through a VSD
♦
Most common cyanotic congenital anomaly
♦
Four classic anatomical findings:
VSD
Dextroposition of the aorta that overrides the VSD and originates from both right
and left ventricles
Right ventricular outflow obstruction (pulmonary or subpulmonary stenosis)
Right ventricular hypertrophy
♦
The degree of cyanosis is related to the severity of obstruction to the pulmonary circulation; increased right ventricular outflow obstruction augments right-to-left shunting
♦
Right-to-left shunt predisposes patients to develop cerebral abscesses or stroke
Transposition of Gr eat Arteries
♦
Approximately 4% of congenital heart defects
♦
Aorta arises from morphologic right ventricle and is situated anterior and to the right of the pulmonary artery (normal is situated posterior and to the right of pulmonary artery); pulmonary artery arises from morphologic left ventricle
♦
To survive, mixing of blood between the two parallel circulations occurs through a PFO, ASD, VSD, or PDA
♦
Cyanosis at birth
Tricuspid Atresia
♦
Right-to-left shunt through a PFO or ASD
♦
Tricuspid valve absent and associated with hypoplasia of right ventricle and pulmonary artery
♦
VSD may allow flow from left ventricle to pulmonary artery
♦
Cyanosis present from birth
Truncus Arteriosus
♦
Right-to-left shunt
♦
Single arterial vessel arises from both ventricles above a VSD
♦
Presents with cyanosis from birth
Ventricular Septal Defect (VSD)
♦
Left-to-right shunt
♦
Most common cardiac defect seen in children, approximately 20% of congenital heart defects
♦
May be divided into (1) small, usually muscular-type defects that spontaneously close in the first few years of life; (2) small, usually perimembranous-type defects with minor symptoms and which do not cause pulmonary hypertension; and (3) large hemodynamically significant defects
♦
Mostly occurs in perimembranous portion of the interventricular septum
♦
May predispose to infective endocarditis, aortic insufficiency if one of the aortic cusps prolapse into the VSD
♦
Large VSDs, if untreated, will produce volume overload and pulmonary hypertension, biventricular hypertrophy, and congestive heart failure and may lead to Eisenmenger syndrome
Acquired Diseases of the Myocardium
Ischemic Heart Disease
♦
Ischemic heart disease is due to an imbalance between coronary perfusion and myocardial oxygen demand. It manifests as diverse clinical ischemic syndromes including stable and unstable angina, myocardial infarction (MI), chronic ischemic heart disease, and sudden cardiac death
Acute Myocardial Infarction
Clinical
♦
Most often due to coronary artery atherosclerosis in over 90% of cases; less often due to coronary vasospasm, coronary artery dissection, coronary thrombosis, or embolism
♦
Plaque fissure or rupture may cause a totally occlusive thrombosis leading to acute ST-segment elevation MI or a nonocclusive thrombosis leading to unstable angina or non-ST-segment elevation MI
♦
Myocardial necrosis occurs in a wavefront phenomenon from the subendocardium to the subepicardial myocardium
♦
Extent of myocardial necrosis depends on length of time of occlusion and degree of collateral blood flow
♦
Complications of large transmural infarcts include:
Congestive heart failure – likely to develop if >40% of left ventricle is infarcted
Arrhythmia – conduction block, bradyarrhythmias, and tachyarrhythmias
Infarct extension/reinfarction – development of new necrosis in the same area of a recent infarction as evidenced by recurrence of chest pain, elevated cardiac enzymes, and electrocardiographic changes
Infarct expansion and left ventricular aneurysm – infarcted zone becomes thin and stretched out, forming a regional left ventricular cavity dilatation
Mural thrombosis/embolism – thrombi can form in expanded akinetic infarcted wall usually in the setting of large anteroapical MI, with risk of systemic embolization
Right ventricular infarction – associated with posterior wall infarction and high pulmonary pressures
Cardiac rupture
•
Free wall rupture leading to hemopericardium, most often in the anterior wall
•
Interventricular septal rupture leading to left-to-right shunt
•
Papillary muscle rupture leading to acute mitral regurgitation
•
Ruptures usually occur in the first 10 days of MI
Pericarditis – may be seen in the area of acute infarction or represent a late postinfarction inflammation (Dressler syndrome) appearing weeks after infarction
Macroscopic
♦
Myocardial necrosis in an acute MI appears grossly as pale yellow areas in the myocardium
♦
If reperfusion has occurred, the infarcted areas may appear red
♦
In the first 6–12 h, usually no grossly detectable changes unless using tetrazolium incubation (Fig. 27.2)
♦
After 18–24 h, there may be either myocardial pallor or mottling
♦
In 2–3 days, the infarcted zone begins to appear yellow as polymorphonuclear leukocytes infiltrate the tissue, and the pallor increases as more polymorphonuclear leukocytes continue to infiltrate the infarcted myocardium (Fig. 27.3)
♦
At 7 days, distinct gelatinous early scar with red borders and depression on cut surface is present
Microscopic
♦
Coagulation necrosis – hypereosinophilia with blurring or loss of the striated pattern of the myocyte sarcoplasm (Fig. 27.8)
♦
Contraction band necrosis (which may be part of reperfusion injury including interstitial hemorrhage) is frequently present (Fig. 27.10)
♦
Wavy and thinned myocytes can also be seen; however, wavy myocytes without thinning should not be interpreted as infarcted myocardium (Fig. 27.11)
♦
♦
If reperfusion occurs, contraction band necrosis is prominent with interstitial hemorrhages
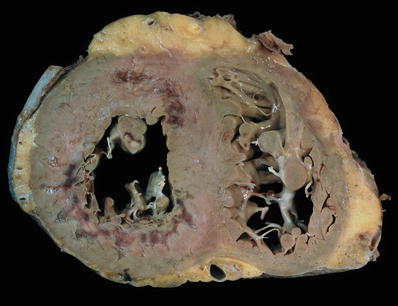
Fig. 27.1.
Cross section of the ventricles showing subendocardial infarcts in the anterior and posterolateral walls that extend into the septum. The infarcts have a gelatinous texture and the red areas represent granulation tissue. Bordering the infarcts, there are subtle areas of white gray discoloration which represent areas of early scar formation. Also note the infarct in the anterolateral papillary muscle.
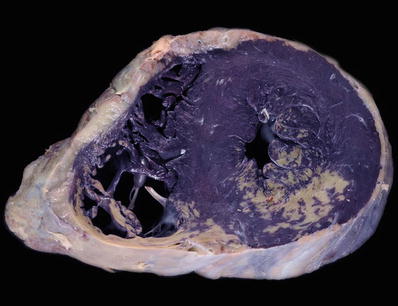
Fig. 27.2.
Cross section of the ventricles fixed in formalin after incubation in nitroblue tetrazolium chloride . Nitroblue tetrazolium is transformed to blue/purple dye by lactic dehydrogenase, indicating viable tissue. This image shows two distinct infarcts (lack of blue/purple staining) in the posterior wall of the left and right ventricles. This stain accurately detects infarcts less than 4 h in evolution, before any reliable histologic finding can be seen.
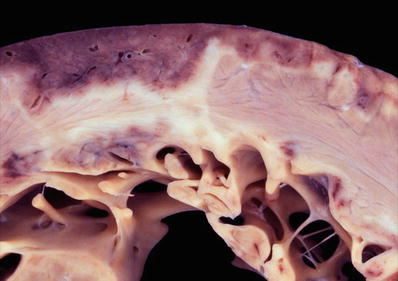
Fig. 27.3.
The myocardiu m shows a yellow demarcation between the viable subepicardial myocardium and infarcted subendocardial myocardium on the left. The infarct becomes transmural on the right side of the field. The paler yellow border represents the zone of maximal infiltration of neutrophils at 2–3 days.
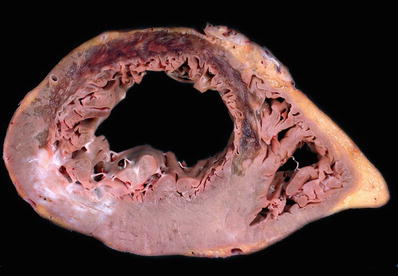
Fig. 27.4.
An extensive transmural anteroseptal left ventricular infarct shows thinning of the myocardium with gelatinous change consistent with early scar formation between 7 and 14 days. Islands of necrotic myocardium may persist in large infarcts as seen in the anterior wall in this case. A healed infarct with white scar is present in the posterolateral wall.
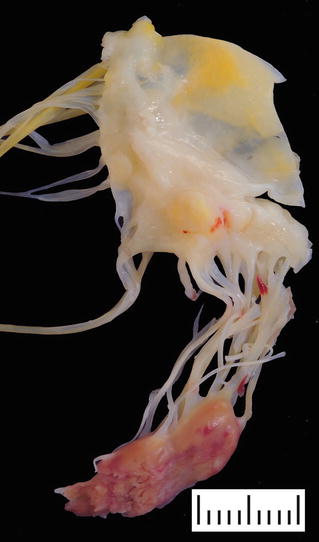
Fig. 27.5.
Surgical specimen showing a segment of mitral valve and a ruptured papillary muscle secondary to myocardial infarction. Note the pale myocardium with hemorrhages and the ragged edges of the papillary muscle head.
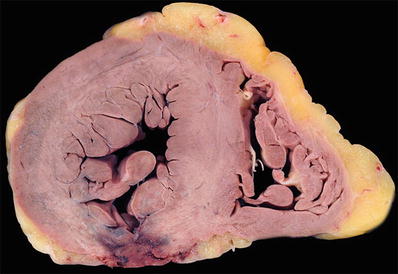
Fig. 27.6.
Acute transmural infarction in the posterior wall evolved into a rupture site. The image shows a serpiginous hemorrhagic path of the blood dissecting through the necrotic myocardium.
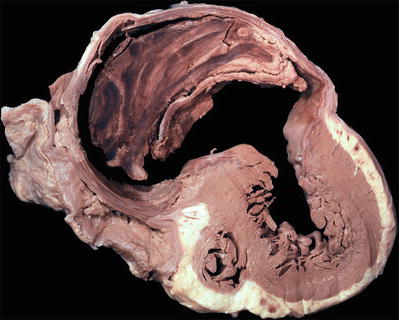
Fig. 27.7.
A pseudoaneurysm with laminated thrombus is shown surrounded by fibrous tissue and pericardium. A pseudoaneurysm results from a contained rupture of the ventricular wall and communicates with the ventricular cavity through a narrow neck. In comparison, a true ventricular aneurysm results from dilatation of the scarred myocardium.
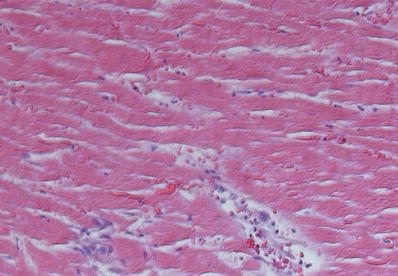
Fig. 27.8.
Coagulation necrosis of the myocardium showing hypereosinophilic sarcoplasm of the myocytes with indistinct or frankly blurred striations and loss of nuclei.
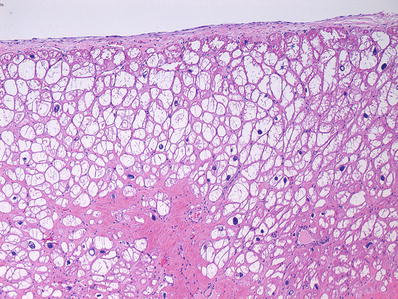
Fig. 27.9.
Colliquative myocytolysis showing large vacuolated sarcoplasm of myocytes due to hydropic change. It usually occurs in subendocardial location. It may also be seen in areas of “hibernating” myocardium in chronic ischemic injury.
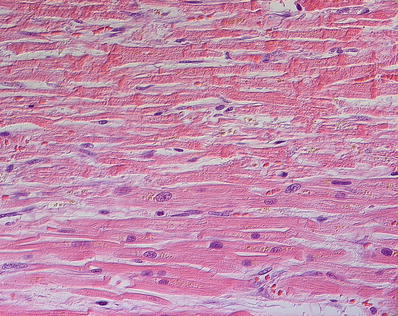
Fig. 27.10.
Contraction band necrosis showing transverse hypereosinophilic bands alternating with pale granular spaces along the length of the myocytes. The transverse bands result from overlapping of hypercontracted sarcomeres within a myocyte. For comparison the myocytes in the lower portion of the field do not show contraction band necrosis.
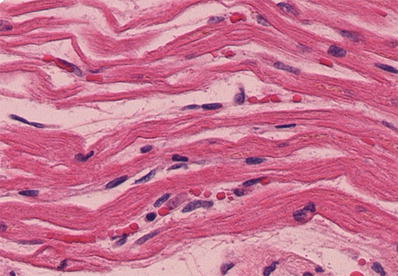
Fig. 27.11.
An early morphologic change in acute myocardial infarction is the appearance of wavy and thinned fibers. Note the capillary congestion in these areas, lack of polymorphonuclear infiltration, and presence of hypereosinophilic fibers in the myocytes.
Table 27.3.
Sequence of Certain Microscopic Changes in Acute Myocardial Ischemia (Approximate)
Time after event
Histopathologic findings
4–6 h
Margination of polymorphonuclear leukocytes (Fig. 27.12)
8–12 h
Diapedesis of polymorphonuclear leukocytes into the myocardial interstitial space (Fig. 27.13)
1 day
Hypereosinophilic fibers, nuclear pyknosis, coagulation necrosis, colliquative myocytolysis, edema, interstitial hemorrhage, contraction band necrosis, increase in neutrophilic infiltration
2–3 days
Marked polymorphonuclear infiltrate with extensive karyorrhexis, loss of myocyte nuclei (Fig. 27.14)
4 days to 1 week
Macrophage infiltration, early granulation tissue with fibroblast response, and capillary proliferation at the edges (Fig. 27.15)
7–14 days
Granulation tissue with hemosiderin-laden macrophages; variable amount of lymphocytes, rare plasma cells, and eosinophils; polymorphonuclear leukocytes have disappeared
2–8 weeks
Granulation tissue matures with increased collagen deposition, which becomes prominent and more dense (Fig. 27.16)
2 months
Healed scar
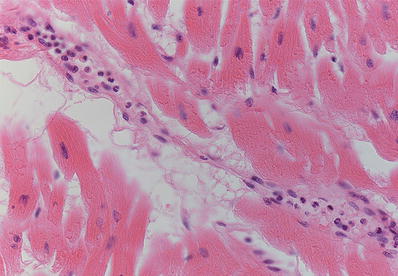
Fig. 27.12.
Margination of polymorphonuclear leukocytes is one of the earliest unambiguous changes in myocardial infarcts. It is seen as early as 4–6 h postinfarction.
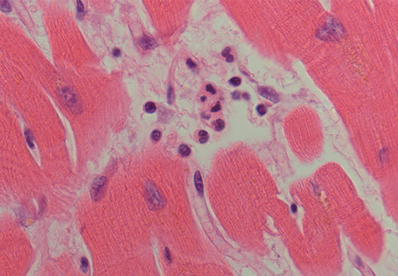
Fig. 27.13.
Once the polymorphonuclear leukocytes marginate inside capillaries near the infarcted area, they begin to diapedese into the extracellular space and infiltrate the surrounding myocardium. This change usually starts at around 6–8 h and increases with time as more polymorphonuclear leukocytes are chemoattracted to the infarcted myocardium.
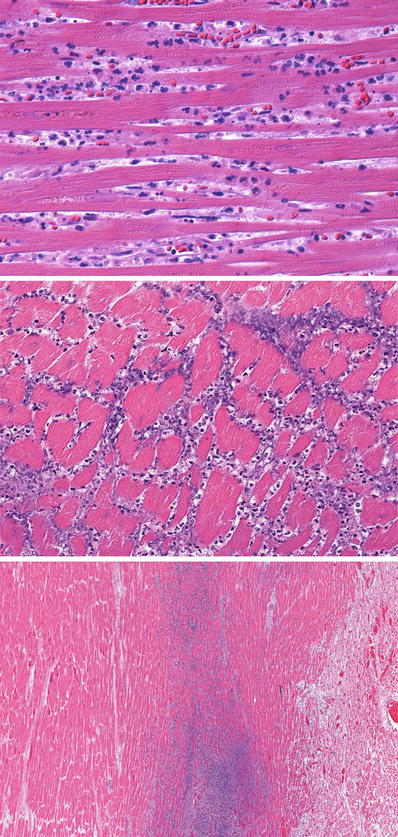
Fig. 27.14.
The top panel shows frank infiltration of the interstitium by further diapedesis of polymorphonuclear leukocytes. The myocardium shows coagulation necrosis. This amount of polymorphonuclear leukocyte infiltration occurs at around 24 h of evolution of the infarct. The nuclei of the myocytes are not staining, but striations can still be identified in the sarcoplasm. The middle panel shows extensive karyorrhexis of the polymorphonuclear leukocytes, which imparts a “dusty” basophilic appearance and is observed at 3–4 days postinfarct. The lower panel shows a zone of basophilia representing polymorphonuclear cells undergoing karyorrhexis between the zone of coagulative necrosis on the left and viable myocardium on the right.
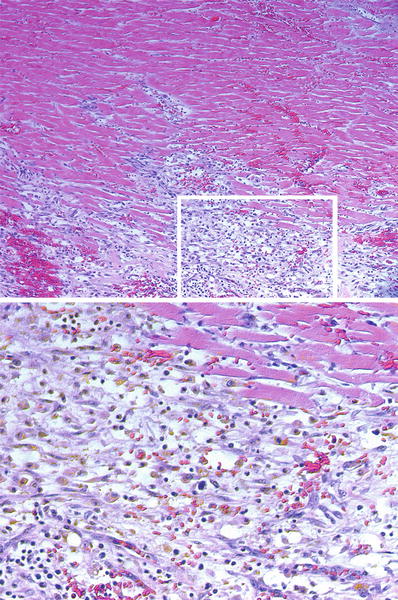
Fig. 27.15.
In the deepest areas of a large infarct, the acute inflammatory infiltrate may not reach and promote lysis of the myocytes. Thus, dead myocytes appear eosinophilic, but do not show nuclei (karyolysis). Similarly the endothelial cells of the capillaries undergo karyolysis. However, as the repair response approaches this core, it shows abundant fibroblasts, and hemosiderin-and-lipofuscin laden macrophages become ubiquitous (inset and lower image). This infarct is about 7 days in evolution.
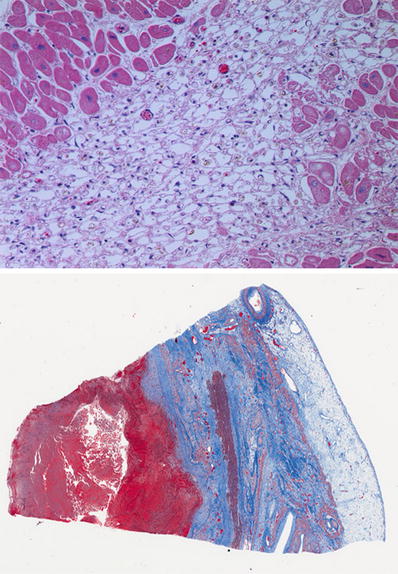
Fig. 27.16.
Another pattern commonly seen is the disappearance of myofibrils from the sarcoplasm, which leaves empty spaces bounded by the sarcolemma resulting in an alveolar pattern with scattered macrophages. This pattern is usually seen in small areas of infarction or in the outer zone of a large infarct. The lower image shows a healing transmural infarct with pale and dark blue zones of collagen deposition. Note the mural thrombus with the red fibrin in a trichrome stain (red).
Chronic Ischemic Heart Disease
Clinical
♦
Insidious onset of congestive heart failure due to progression of myocardial ischemic damage following previous MIs
Macroscopic
♦
Chamber dilatation and hypertrophy
♦
Patchy endocardial fibrosis
♦
Severe atherosclerosis of coronary arteries
Microscopic
♦
Colliquative myocytolysis – hydropic change in myocytes, most prominent in the subendocardial myocardium
♦
Interstitial and replacement fibrosis
♦
Compensatory myocyte hypertrophy
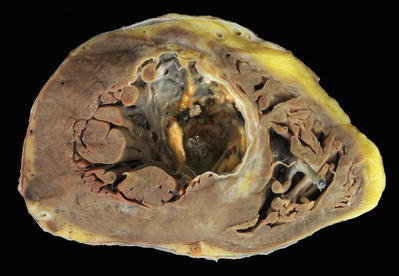
Fig. 27.17.
A healed transmural infarct of the anterior septum and a small portion of the anterior left ventricular wall appear as dense white scar with focal calcification. There is an apical thrombus. Note the fine areas of gray white fibrosis in the noninfarcted myocardium. The right ventricle shows a segment of a pacing lead with a fibrous tissue cuff surrounding it.
Cardiomyopathy
♦
Cardiomyopathies are a heterogeneous group of diseases of the myocardium associated with mechanical and/or electrical dysfunction that usually (but not invariably) exhibit inappropriate ventricular hypertrophy or dilatation
♦
There is absence of coronary artery disease, valvular disease, congenital heart disease, and hypertension sufficient enough to explain the myocardial disorder
♦
Etiology is varied but frequently is genetic
♦
Cardiac involvement can be the primary manifestation of the disease (primary cardiomyopathies), or it may be part of generalized systemic disorders (secondary cardiomyopathies)
♦
The pathology of cardiomyopathies can be classified in three major practical categories which correlate the morphology and functional phenotype: dilated, hypertrophic, and restrictive as shown in Table 27.4
Table 27.4.
Morphologic and Clinical Physiologic Classification of Cardiomyopathies
Functional pattern | LV ejection fraction (%) | Mechanisms of failure | Causes of phenotype | Differential diagnosis: indirect myocardial dysfunction (mimicking cardiomyopathy) |
|---|---|---|---|---|
Dilated | <40 | Impaired contractility (systolic dysfunction) | Genetic, inflammatory, toxic, idiopathic | Ischemic, valvular, hypertensive, congenital heart disease |
Hypertrophic | 50–80 | Impairment of compliance (diastolic dysfunction) | Genetic, Friedreich ataxia, storage diseases, infants of diabetic mother | Hypertensive heart disease, aortic stenosis |
Restrictive | 45–90 | Impairment of compliance (diastolic dysfunction) | Amyloidosis, radiation-induced fibrosis, idiopathic | Pericardial constriction |
♦
Each phenotype can be further classified into familial/genetic or nonfamilial/nongenetic in etiology
Dilated Cardiomyopathy
Clinical
♦
Ca n occur at any age
♦
Signs and symptoms of systolic heart failure – dyspnea, orthopnea, fatigue; evidence of low cardiac output on examination including hypotension, tachycardia, cool extremities, mental status changes; evidence of volume overload including weight gain, peripheral edema, jugular venous distension
♦
Genetic in at least 30% of cases
♦
Phenotype associated with pregnancy or the postpartum period (peripartum cardiomyopathy), alcoholism (alcoholic cardiomyopathy), myocarditis, muscular dystrophies, catecholamine excess, takotsubo “stress cardiomyopathy”
Macroscopic
♦
Cardiomegaly with left ventricular cavity dilatation, often with four-chamber dilatation (Fig. 27.18)
♦
Left ventricular wall thickness is increased but may be near normal as the hypertrophy is masked by chamber dilatation
♦
Valvula r annulus dilated
♦
Mu ral thrombi can be present in the atrial appendages and ventricles
♦
Absence of significant coronary artery disease
Microscopic
♦
Histologic changes are usually nonspecific as to the etiology of dilated cardiomyopathy
♦
Variable d egrees of myocyte hypertrophy, degeneration, and interstitial fibrosis
♦
Inflammatory infiltrates minimal and confined to areas of interstitial fibrosis
♦
Myocyte sarcoplasm vacuolization secondary to toxic drugs or other chemicals
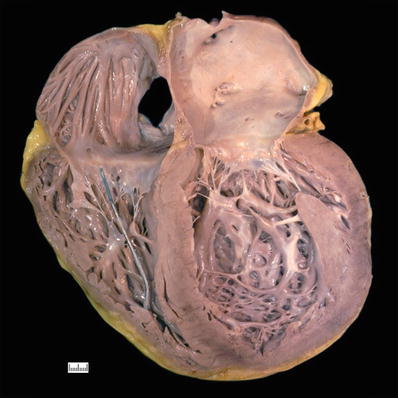
Fig. 27.18.
A case of dilated cardiomyopathy with enlargement of all four chambers, most severe in the left ventricle which appears globular. The wall thickness may be normal as the hypertrophy is masked by the dilatation. The right ventricle shows a segment of a pacing lead well anchored in the apex of this chamber.
Hypertrophic Cardiomyopathy
Clinical
♦
Left ventricu lar hypertrophy in the absence of hypertension and aortic valve disease
♦
50% familial (autosomal dominant), genes encoding sarcomeric proteins mutated
♦
Mutations in β-myosin heavy chain, myosin-binding protein C, and troponin T account for up to 80% of cases with genetic mutations
♦
Symptoms of left ventricular outflow obstruction in 25% of patients
♦
Associated with sudden death during exercise
♦
Treated by beta adrenergic blockage; septal myectomy if with obstructive physiology
Macroscopic
♦
If obstructive, mitral valve thickening and focal endocardial thickening in the outflow tract as a result of contact with anterior mitral leaflet during systole
♦
Enlarged left at rium
Microscopic
♦
Variable fibrosis (interstitial and replacement type)
Differential Diagnosis
♦
Amylo idosis, Fabry disease, storage diseases, mitochondrial disorders, Friedreich ataxia
♦
Hypertensive or valvular (especially aortic) heart disease, which may show concentric left ventricular hypertroph y
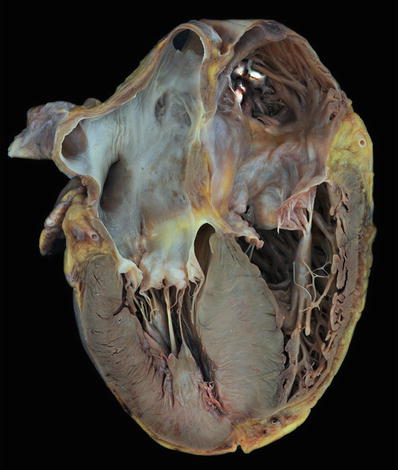
Fig. 27.19.
This heart shows marked hypertrophy of the interventricular septum which is twice as thick as the left ventricular free wall. There is dilatation of the other chambers as well with a white organizing thrombus in the right atrial appendage.
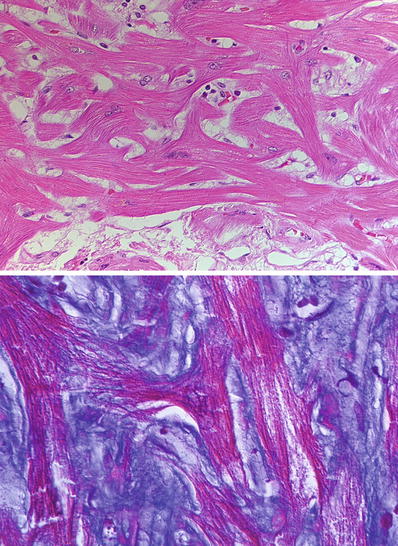
Fig. 27.20.
In hypertrophic cardiomyopathy, the hallmark of the disease is “disarray.” Disarray occurs at the fascicle level, myocyte level, and sarcomere level. At the myocyte level, the myocyte sarcoplasm is disorganized forming branches in contrast to a normal parallel arrangement in sections taken from the interventricular septum. The disarray is also evident in the myofibrils within individual myocytes. This is often accompanied by interstitial fibrosis as shown in the trichrome stain.
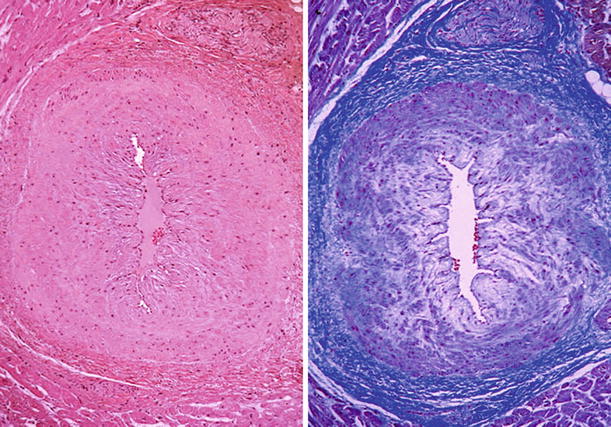
Fig. 27.21.
The intramural coronary arteries in the septum of hearts with hypertrophic cardiomyopathy are often abnormal. The lumen is narrowed, and the wall is thickened by an increase in the smooth muscle cells and ground substance in the media.
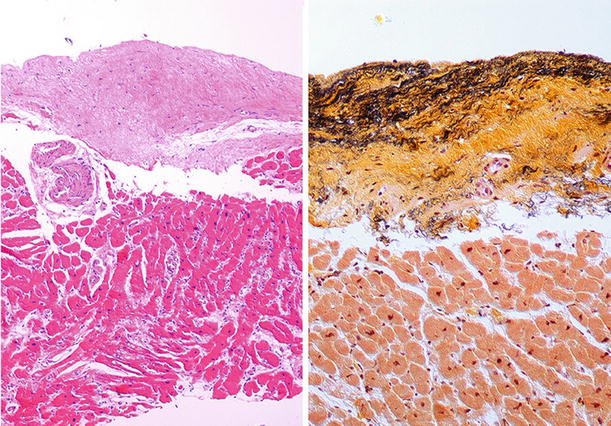
Fig. 27.22.
The outflow tract of the left ventricle in the obstructive type of hypertrophic cardiomyopathy shows endocardial thickening with fibrosis and elastosis evident in the Movat stain.
Arrhythmogenic Right Ventricular Cardiomyopathy
Clinical
♦
Global or regional dysfunction of the right ventricle that may progress to involve the left ventricle
♦
Associated with arrhythmias of right ventricular origin, heart failure, and sudden death
♦
Mutations in desmosomal protein-encoding genes (plakoglobin, plakophilin-2, desmoplakin, desmocollin-2, desmoglein-2) are common; other mutations involving transmembrane protein 43, transforming growth factor-beta 3, desmin, and titin are reported
Macroscopic
♦
♦
Right ventricular dilatation and wall thinning
♦
Left ventricle may also be involved by the fibrofatty infiltration and replacement
Microscopic
♦
Myocyte loss in the compact zone with transmural replacement by adipose and/or fibrous tissue (Fig. 27.26)
♦
Mononuclear leukocytic infiltrates can be present
Differential Diagnosis
♦
Normal adipose tissue infiltration of right ventricle, especially in obese persons, is usually focal without fibrosis and without complete replacement of compact myocardium
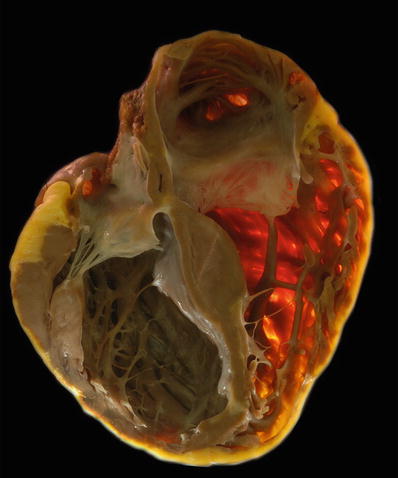
Fig. 27.23.
Transilluminated specimen shows loss of the compact zone in the right ventricle which appears translucent in a case of arrhythmogenic right ventricular cardiomyopathy. Also note the thinning of the left ventricular wall and interventricular septum toward the apex due to fibrofatty replacement.
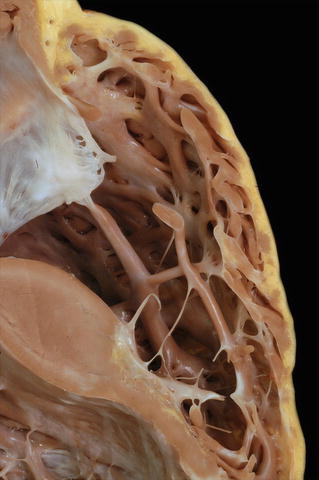
Fig. 27.24.
The right ventricular wall of the same heart in Fig. 27.23 is shown. The compact zone is discontinuous with fatty infiltration and fibrous replacement. The trabecular myocardium is relatively spared.
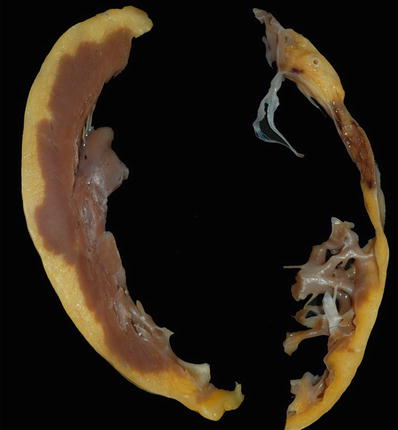
Fig. 27.25.
In addition to the marked thinning of the wall of the right ventricle (shown on the right) due to fibrofatty replacement in arrhythmogenic right ventricular cardiomyopathy, involvement of the left ventricle with fatty infiltration producing a “moth-eaten” appearance is seen in almost half of the cases. Note the irregular contour of the subepicardium of the left ventricle in this example.
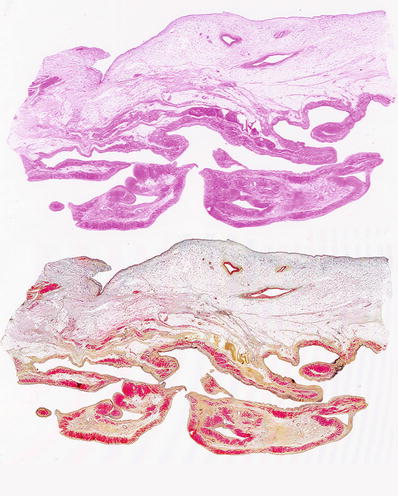
Fig. 27.26.
Section of the right ventricular wall with extensive adipose tissue replacement of the compact zone and trabecular myocardium as shown in an H&E stain and corresponding Movat stain. The Movat stain highlights the fibrous tissue in yellow.
Restrictive Cardiomyopathy
Clinical
♦
Diastolic dysfunction with impaired relaxation (must be distinguished from constriction)
♦
Least common type of the cardiomyopathies
♦
Could be idiopathic or secondary due to stiff myocardium or thickened endocardium
♦
E ndocardium-based restriction:
Endomyocardial fibrosis
Diffuse endocardial fibroelastosis
Loeffler (hypereosinophilic) endomyocarditis
♦
Myocardial interstitium-based restriction:
Amyloidosis (see below under Amyloidosis)
Postradiation fibrosis
Fibrosing sarcoidosis
Macroscopic
♦
The macroscopic features vary depending on the etiology of the restriction
♦
Hea rt size normal or only slightly enlarged
♦
Ventricular cavities normal or mildly dilated; atrial cavities moderately to severely dilated
♦
Lef t ventricular wall thickness may be normal
♦
Endocardial fibrosis that may obliterate the apices of the right and left ventricles and surround the papillary muscles
♦
Mural thrombi may be present in endomyocardial fibrosis
Microscopic
♦
Primary restrictive cardiomyopathy shows myocyte hypertrophy and interstitial fibrosis
♦
Severe fibrous thickening of the endocardium with or without eosinophils and organizing thrombi
♦
Elastic fiber proliferation in endocardial fibroelastosis seen in the pediatric age group often secondary to aortic steno sis
Infiltrative Myocardial Diseases
Amyloidosis
Clinical
♦
Depending upon the type and degree of involvement, it may be asymptomatic or present with congestive heart failure, arrhythmias, ischemic disease, and sudden death
♦
Light chains (kappa or lambda) are the most common amyloidogenic proteins (60%) found in the heart, followed by transthyretin (TTR) (40%) in symptomatic patients
♦
Senile amyloidosis may be due to TTR or less commonly atrial natriuretic factor/peptide (ANF/ANP)
Macroscopic
♦
Extensive deposits may lead to pale, firm, and rubbery myocardium
♦
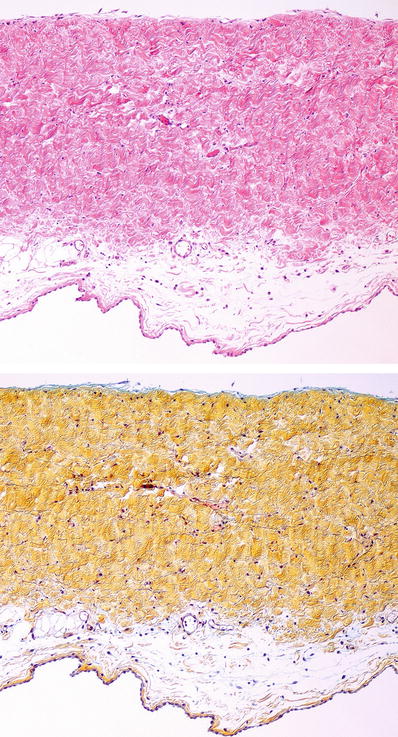
Left atrial endocardial and valvular deposits may appear waxy and shiny yellow/ochre fine nodules (Fig. 27.27)
Microscopic
♦
Extracellular, eosinophilic homogeneous (also called “amorphous”) material on hematoxylin and eosin (H&E)-stained section (Fig. 27.28)
♦
Deposits can be interstitial surrounding myocytes or nodular
♦
Vascular wall involvement can be present, most commonly in light-chain amyloidosis, and may cause vessel stenosis
♦
By definition, deposits are Congo red positive, with apple-green birefringence when examined with polarized light microscopy (Fig. 27.29)
♦
Alternatively, sulfated Alcian blue stain has been used However, thioflavin-T and thioflavin-S stains are very sensitive but require UV light microscopy
♦
Immunohistochemical identification of cardiac amyloid is specific for the common subtypes that affect the heart (kappa light chains, lambda light chains, transthyretin, atrial natriuretic factor) (Fig. 27.30). Other subtypes may need mass spectroscopy for identification
♦
Electron microscopy is also definitive (showing 10 nm extracellular fibrils) but rarely necessar y
Glycogen Storage Diseases
♦
Excess sequestration of various glycogen storage products in lysosomes or free in the sarcoplasm leads to heart failure
Hemochromatosis/Hemosiderosis
♦
Systemic iron deposition with organ damage (usually in hemochromatosis)
♦
Morphology alone cannot differentiate hemochromatosis from hemosiderosis
Angiokeratoma Corporis Diffusum Universale ( Fabry Disease )
♦
X-linked recessive inheritance
♦
Deficiency of lysosomal alpha-galactosidase leading to ceramide trihexoside accumulation
♦
Skin, cornea, kidney, and heart affected
♦
Intralysosomal concentric or parallel lamellae (myelin figures) by electron microscopy are the hallmark (Fig. 27.34)
♦
Differential diagnosis: chloroquine and hydroxychloroquine cardiotoxicity also shows myocyte vacuolization and presence of myelin figures; clinical history is very important to distinguish between these two entities
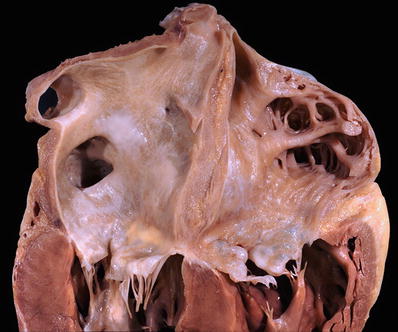
Fig. 27.27.
In extensive cardiac amyloidosis, the atria are enlarged, and the endocardium of both atria shows fine yellow-ochre granular surface due to amyloid deposition. These deposits are also seen in the tricuspid and mitral valve leaflets. The ventricular myocardium shows subtle areas of pallor which correspond to amyloid deposits.
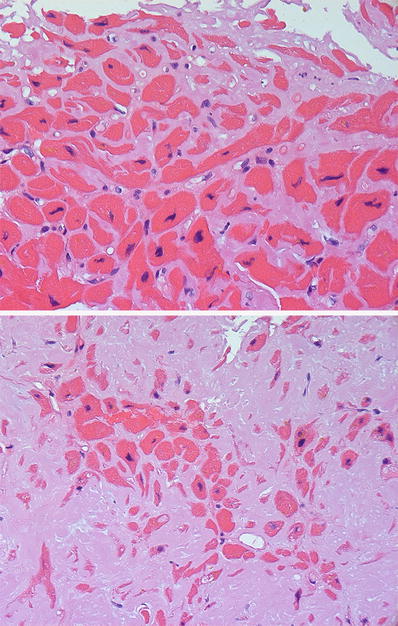
Fig. 27.28.
Amyloid infiltration in the heart shows “amorphous” eosinophilic material accumulating in the extracellular space. The amyloid is deposited throughout the interstitium surrounding individual myocytes (top panel). In advanced disease, there is more pronounced myocyte atrophy with accumulation of the interstitial deposits into a nodular pattern (bottom panel).
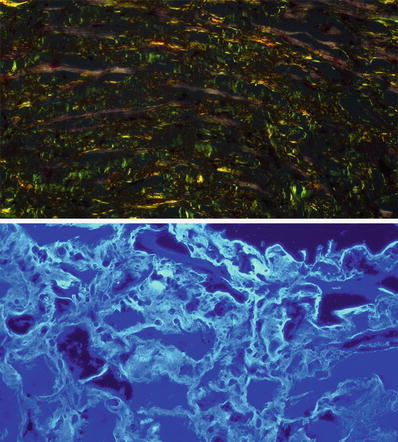
Fig. 27.29.
Interstitial amyloid shows apple-green birefringence on polarization microscopy (top panel) when stained with Congo red. Amyloid deposits also appear fluorescent with thioflavin-S or thioflavin-T staining viewed under fluorescence microscopy. This is a more sensitive and reproducible stain than Congo red.
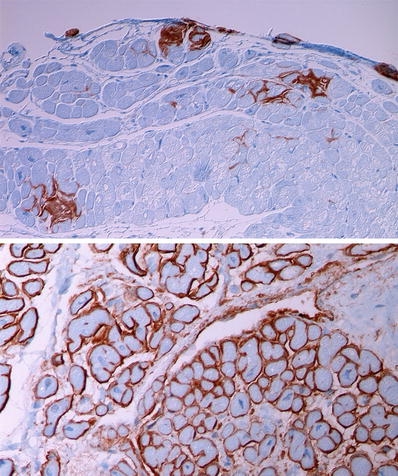
Fig. 27.30.
Immunohistochemical staining is useful for typing of cardiac amyloidosis. The top panel shows focal deposits of transthyretin in a coarse interstitial pattern and forming small nodules. The bottom panel shows a diffuse interstitial perimyocytic pattern of deposition in light-chain amyloidosis where lambda light chains are at least twice as frequently seen compared to kappa light-chain deposition.
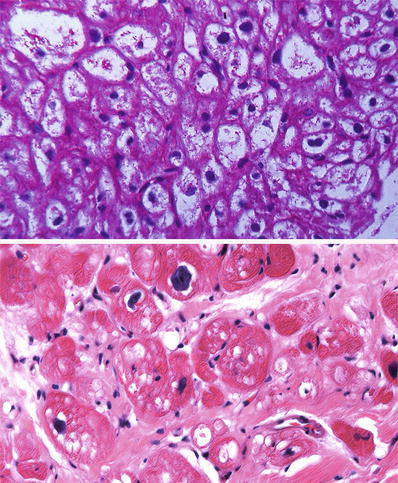
Fig. 27.31.
The myocytes are enlarged with pale sarcoplasm due to massive accumulation of glycogen that stain PAS positive in a case of Pompe disease (top panel). In the adult, glycogen storage disease may appear as irregular vacuoles associated with interstitial fibrosis (bottom panel).
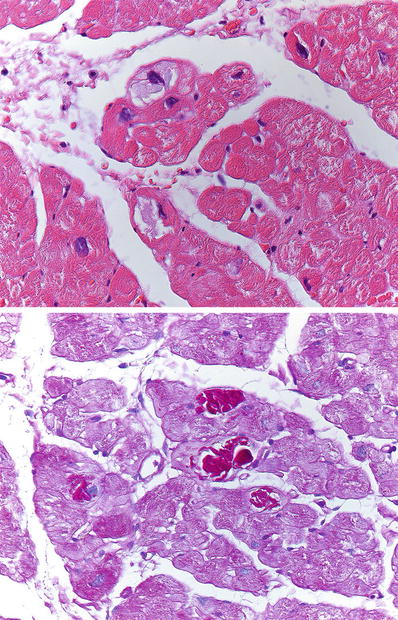
Fig. 27.32.
A few myocytes show accumulation of basophilic material in the sarcoplasm that is intensely positive with PAS. This type of glycogen deposition can be seen in type IV glycogen storage disease and in hearts of patients older than 65 years of age (basophilic degeneration).
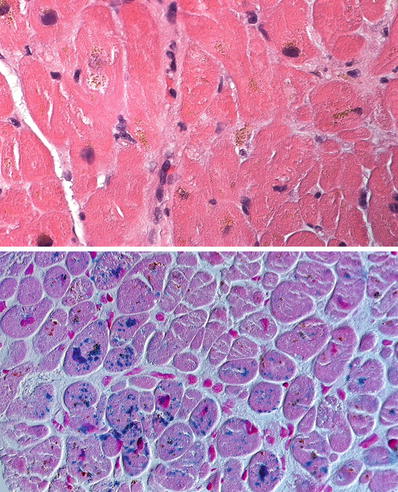
Fig. 27.33.
The myocytes contain dark yellow-to-brown granular deposits mostly in perinuclear location (top panel) which are readily identified as iron on a Prussian blue stain (bottom panel) in a case of hemosiderosis.
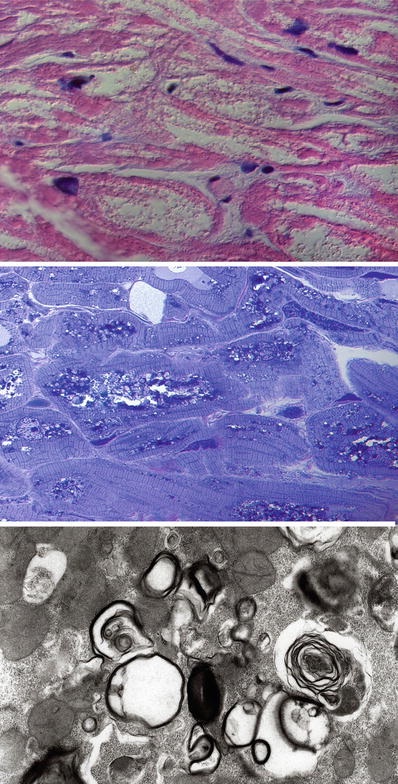
Fig. 27.34.
In Fabry disease, there is marked vacuolation of the myocytes. The myofibrils are displaced to the periphery by the deposits occupying the central clear to finely granular area (top panel). On toluidine blue stain of a semithin section, the glycolipid deposits are evident as dark blue metachromatic deposits. Ultrastructural examination demonstrates that the metachromatic deposits are in fact the characteristic lamellar bodies seen in Fabry disease.
Myocarditis
Clinical
♦
Associated with viral infections (most often coxsackie, echovirus, influenza, adenovirus, parvovirus B19), autoimmune diseases, and drug reactions
♦
Bacterial, fungal, protozoal, and parasitic myocarditis are far less common
♦
Complications include congestive heart failure, conduction defects, arrhythmias, and sudden death
Macroscopic
♦
Range from normal to biventricular dilatation/hypertrophy with pale “flabby” myocardium and fibrosis
Microscopic
♦
Dallas criteria for the evaluation of myocarditis in biopsies: leukocytic infiltrate (usually lymphocytic) with myocyte degeneration/necrosis
♦
In most cases, a T-cell lymphocytic infiltrate admixed with histiocytes (Fig. 27.35), occasionally with eosinophils
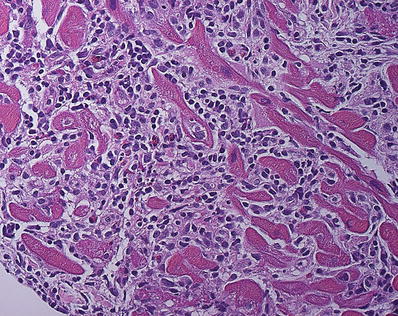
Fig. 27.35.
Lymphocytic myocarditis showing dense infiltrates of lymphocytes, histiocytes, and few eosinophils that expands the interstitial space. Myocyte injury is evident in the left lower corner of the field showing fragmentation and irregular borders of the myocytes.
♦
Myocyte injury can be spotty or geographic
Giant Cell Myocarditis
Clinical
♦
Occurs in young and middle-aged adults
♦
Presents with arrhythmias, conduction defects, cardiac failure, and sudden death (50%)
♦
Rapidly progressive and fatal; heart transplantation may be indicated
♦
Associated with thymoma, lupus, and inflammatory bowel disease
Microscopic
♦
Geographic myocyte necrosis with lymphohistiocytic infiltration, eosinophils, and plasma cells (Fig. 27.36)
♦
Lacks discrete granulomas
♦
Giant cells of both macrophage and myocyte origin are present
Differential Diagnosis
♦
Cardiac sarcoidosis
Cardiac involvement in sarcoidosis manifests as arrhythmias, congestive heart failure, and sudden death
On gross examination, sarcoidosis granulomata occur in the interventricular septum toward the base and spread toward the lateral walls of the right and left ventricle
The granulomata and the destroyed myocardium are replaced by dense fibrous tissue, producing firm to rubbery white to gray and glassy scars (Fig. 27.37)
Myocardial dropout associated with discrete nonnecrotizing epithelioid granulomata with lymphocytes and eosinophils in the acute stages
Once the myocardium is replaced by fibrous tissue, occasional granulomata remain (Fig. 27.38)
Eosinophilic Myocarditis
♦
Most often due to drug hypersensitivity reaction, less commonly due to hypereosinophilic syndrome, eosinophilic granulomatosis with polyangiitis (Churg–Strauss syndrome), and infection
♦
Eosinophil-rich infiltrate admixed with lymphocytes and histiocytes, commonly in perivascular location
♦
Myocyte necrosis rare and minima l
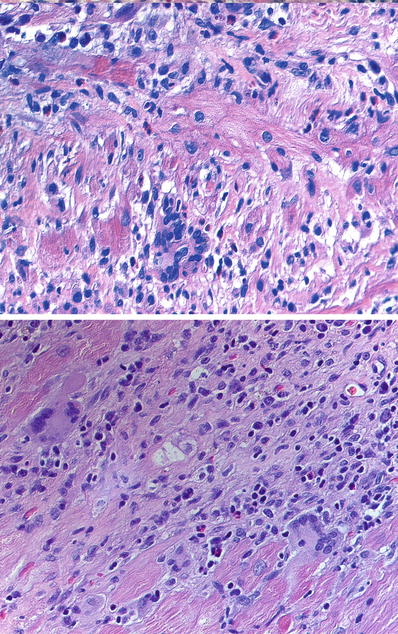
Fig. 27.36.
Giant cell myocarditis showing an aggressive inflammatory infiltrate notable for the presence of multinucleated giant cells and variable amount of eosinophils in both images. Well-formed granulomas are absent.
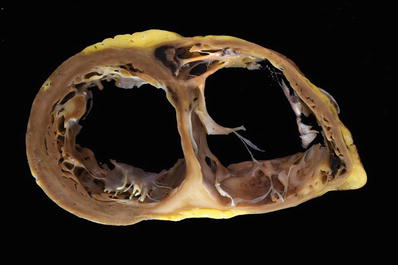
Fig. 27.37.
A case of cardiac sarcoidosis with marked biventricular dilatation of the heart. There is sclerotic white “waxy” appearing formation of scars in the interventricular septum and posterior right ventricular wall. Mural thrombi are present in the right ventricle.
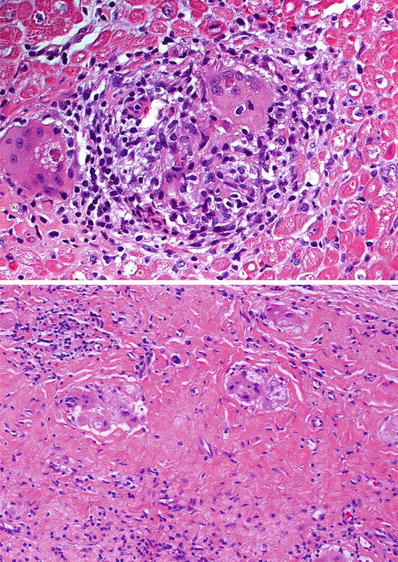
Fig. 27.38.
Cardiac sarcoidosis showing discrete noncaseating granuloma in the myocardium (top panel). Small granulomas and multinucleated giant cells typically persist within dense fibrosis in areas of gross scarring (bottom panel).
Anthracycline Cardiotoxicity
♦
Includes cases associated with doxorubicin (adriamycin) and daunorubicin cardiotoxicity
♦
Earliest changes include myocyte vacuolization secondary to sarcoplasmic reticulum dilatation
♦
EM findings include sarcoplasmic vacuolization, sarcoplasmic reticulum system dilatation, and lysis of myofibrils
♦
Grading (0–3) is based on the percentage of cells affected in ten plastic-embedded blocks of tissue
Hypertensive Heart Disease
♦
Secondary to longstanding systemic hypertension affecting the left ventricle
♦
Grossly, concentric left ventricular hypertrophy (Fig. 27.39)
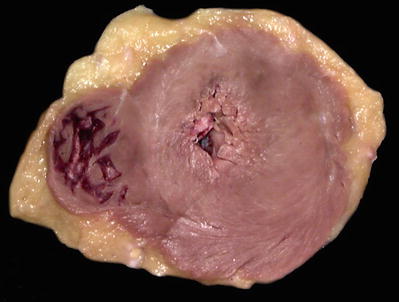
Fig. 27.39.
Concentric left ventricular hypertrophy with a very small cavity.
♦
Must exclude the possible contribution of valvular and myocardial diseases to the hypertrophy
♦
If pulmonary hypertension is the cause, then right ventricular hypertrophy and dilatation occur
Diseases of the Pericardium
Normal Pericardium
♦
Morphologically a parietal pericardium (the sac of fibrous tissue around the heart) and visceral pericardium can be recognized as distinct structures
♦
Both are lined by mesothelial cells, thus forming a serosal layer that is continuous between the parietal pericardium and the visceral pericardium
♦
The mesothelial cell lining produces and reabsorbs pericardial fluid (Fig. 27.40)