Fig. 8.1 Action potentials show variations in patterns among different populations of myocytes in different regions of the heart. The patterns are determined by the opening and closing of selective gates for Na+, Ca2+ and K+, and this figure should be studied in conjunction with Figure 8.2. The overall stability of the resting transmembrane ionic balance is controlled by active pumps, such as the Na+/K+-ATPase pump, which maintain the Na+ concentration gradient of 140 mM outside the cell versus 10–15 mM inside, and the K+ concentration gradient of 140 mM inside and 4 mM outside the cell. This results in an electrical potential at rest of approximately −70 to −80 mV inside the cell relative to 0 mV outside the cell. Large ion fluxes at rest are prevented by specific pumps and closure of voltage-gated gates. Action potentials in the atrioventricular (AV) node, bundle of His and ventricle are controlled by the sinoatrial (SA) node when the heart is in sinus rhythm. The rate of spontaneous depolarisation of the SA node determines its primacy as a pacemaker in the healthy heart. Phase 0 (B, C) occurs when the membrane potential reaches a defined threshold potential and an ‘all-or-none’ influx of Na+ through voltage-dependent fast Na+ channels occurs; this is transient and the gates close after a few milliseconds. Phase 0 is much slower in the SA and AV nodes than in ventricular cells, and depends mainly upon Ca2+ influx (A). This causes the conduction velocity in the SA node to be considerably less than that in the Purkinje fibres, and the refractory period is longer in proportion to the total duration of the action potential. Phase 1, called the early repolarisation and notch, results from K+ efflux (the transient outward (Ito) current) and reduced Na+ influx (Fig. 8.2). The phase 2 plateau is primarily a result of Ca2+ influx (slow inward, or SI, current) which is balanced by K+ efflux over a slow time course. Phase 3 repolarisation results from inactivation of Ca2+ influx and increasing K+ efflux via a number of currents (see text, Table 8.1 and Fig. 8.2). Part of the overall importance of the K+ currents is to maintain a stable resting membrane potential. Phase 4 (A, B) is termed the diastolic or pacemaker depolarisation generated on hyperpolarisation. The phase 4 inward ‘funny’ current (If) involves Na+ and K+ and is gated both by changes in voltage and by cAMP. If controls the rate of spontaneous beating of the heart and is regulated by the sympathetic and parasympathetic nervous systems. Ca2+ currents may also be involved in pacemaker activity in Phase 4. RP, absolute refractory period; RRP, relative refractory period.
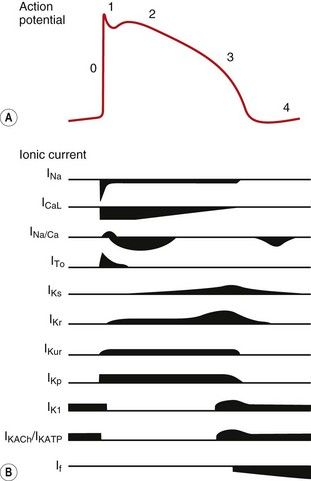
Fig. 8.2 A schematic representation of the influx and efflux of Na+, Ca2+ and K+ in Purkinje fibres. This figure should be examined in conjunction with Table 8.1, with other explanations given in the text. A downward inflection represents influx of the ion, and upward represents efflux. (Modified and reproduced with permission from Tamargo et al. 2004.)
When activated, depolarisation of these cells generates an action potential. The sinoatrial (SA) node, atrioventricular (AV) node, bundle of His and Purkinje system are part of the specialised conducting system of the heart that ensures the depolarizing impulse is conducted rapidly and synchronously through ventricular myocardium. In addition, the AV node slows impulse conduction to allow time for completion of mechanical events in the atrium and for ventricular filling to be completed. Once the impulse is passed to myocytes, depolarisation initiates contraction and also triggers depolarisation in adjacent myocytes. This ensures conduction of the impulse progressively through the myocardium.
The action potentials in cells of the sinoatrial (SA) and atrioventricular (AV) nodes, Purkinje fibres and ventricular muscle vary substantially in their characteristics (Figs 8.1, 8.2 and see Fig. 8.5). Action potentials in cardiac cells can be divided into four phases (Figs 8.1A and 8.2), although phases 1 and 2 are not clearly evident in the SA and AV nodes because of the different ion channels that are activated (Fig. 8.1A).
Cells with pacemaker activity
The myocardial cells of the specialised conducting system (SA node, AV node, bundle of His and Purkinje system) are distinguished electrophysiologically from cardiac myocytes by their intrinsic ability to depolarise spontaneously in phase 4, and independently generate an action potential. These cells are said to possess automaticity and are termed pacemaker cells. Cardiac myocytes have a stable resting potential and do not show phase 4 spontaneous depolarisation (compare Fig. 8.1A and 8.1C).
The primary pacemaker that drives normal repetitive cardiac contractions is the SA node (producing a normal sinus rhythm of 60–100 impulses per minute at rest). These secondary pacemaker, the AV node, depolarises more slowly and can generate 40–60 impulses per minute, while the tertiary pacemakers (the bundle of His, its branches and the Purkinje fibres) can fire 20–40 times per minute. The secondary and tertiary pacemakers will only be utilised if there is a failure of pacemakers that have a faster rate of spontaneous depolarisation.
Control of cell depolarisation in pacemaker and non-pacemaker cells
Slow spontaneous depolarisation in phase 4 in all pacemaker cells results from influx of positive ions (Na+ and K+) into the cell through f-channels that generate the strangely termed cardiac pacemaker ‘funny’ current (If) (Figs 8.1A and 8.2). The If is unusual in being generated by mixed-ion transport through a single channel, in being activated by the negative intracellular potential in diastole and in having slow activation and deactivation rates. Non-pacemaker cells have a minimal or no funny current.
The intrinsic rate of firing of a pacemaker cell depends on four factors:




Activation of the If in the SA node is modulated by the autonomic nervous system via intracellular cAMP. Stimulation of β1-adrenoceptors generates cAMP and shifts activation of the channel to a less negative intracellular voltage, while vagal stimulation via muscarinic M2 receptors inhibits cAMP production and then activation of the channel requires a more negative intracellular voltage.
Full depolarisation of pacemaker cells occurs when, as a result of the If, the internal membrane potential reaches a threshold potential that opens voltage-gated L-type Ca2+ channels so that Ca2+ influx into the cell occurs (phase 0) (Fig. 8.1A; ICaL in Fig. 8.2). The SA node is the dominant pacemaker because activation of the If current in the SA node occurs at a less negative intracellular potential and has a faster intrinsic activation rate than in the AV node or Purkinje fibres. Therefore, spontaneous diastolic depolarisation in the SA node is initiated earlier and the threshold potential for full depolarisation is reached sooner.
Slow influx of Ca2+ in phase 0 is also the reason for slow conduction of the impulse through the AV node. In contrast, phase 0 of the action potential in Purkinje fibres and in atrial and ventricular myocytes is initiated by a rapid influx of Na+ through voltage-gated Na+ ion channels (fast Na+ channels) (Fig. 8.1B; INa in Fig. 8.2).
At the end of phase 0, the intracellular voltage potential briefly becomes positive, at which point a voltage-triggered ‘gate’ closes and inactivates the Na+ or Ca2+ channels and prevents further depolarisation (Fig. 8.1a,b).
Control of cell repolarisation in pacemaker and non-pacemaker cells
Once depolarised, cardiac cells then undergo a process of repolarisation to return the membrane potential to its resting level. This creates the conditions for the next action potential to be initiated (Figs 8.1 and 8.2). In both pacemaker and non-pacemaker cells, repolarisation (phase 3) is achieved by the opening of cell membrane K+ channels known as rectifiers (see Table 8.1 for explanation). In the Purkinje system and non-pacemaker cardiac cells, repolarisation is delayed by influx of Ca2+ through L-type channels (phase 2, the plateau phase, ICaL in Fig. 8.2), which balances K+ efflux (Fig. 8.1B, phase 2). Eventually in these cells the K+ current dominates and the cell returns to a negative intracellular resting potential.
Table 8.1
Selected examples of K+ channels and associated currents
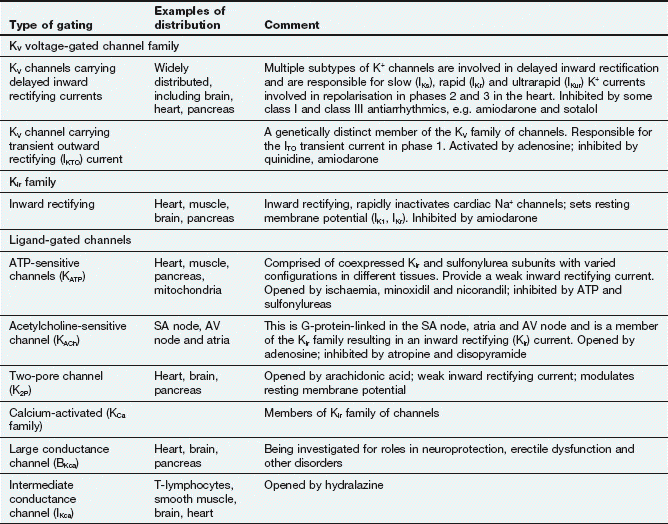
This table should be studied in conjunction with Fig. 8.2.
Potassium channels are diverse in structure and behaviour. Each channel consists of four membrane-spanning subunits, with each subunit consisting of two to six linked membrane segments which make up the water-filled pore. This allows many different configurations of K+ channels, many of which have particular physiological roles. Channels with subunit variations are associated with different types of current which are involved in repolarisation at different phases of the cardiac action potential. The channels can be open or closed depending upon the voltage across the cell or the presence of a selective ligand.
Rectifying current: an inward rectifying current means that under conditions of equivalent but opposing electrochemical potentials these channels pass more current inwards than outwards. An outward rectifying current is similar but in an outward direction.
In the resting phase between action potentials, Na+ and K+ transmembrane concentration gradients are restored by a separate exchange pump (Na+/K+-ATPase; see Fig. 7.4), and intracellular Ca2+ concentration is restored by a Na+/Ca2+ exchange pump. The negative internal resting membrane potential is maintained by high K+ permeability of resting cell membranes through inward rectifying voltage- and ligand-gated K+ channels which close when the cell depolarises (see also Ch. 1).
During the period between phase 0 and the end of phase 2 of the action potential the myocardial cell is refractory to further depolarisation (the absolute refractory period, RP). This is because the depolarising channels are inactivated until a sufficiently negative potential is restored inside the cell. During phase 3, a large depolarising stimulus can open sufficient Na+ or Ca2+ channels (many of which will have recovered to the resting state) to overcome the K+ efflux and initiate a further action potential. This part of the action potential is the relative refractory period (RRP; Fig. 8.1B).
The sum of the individual electrical currents that pass from one cell to another through the heart can be recorded as the surface electrocardiogram (ECG) (Fig. 8.3).
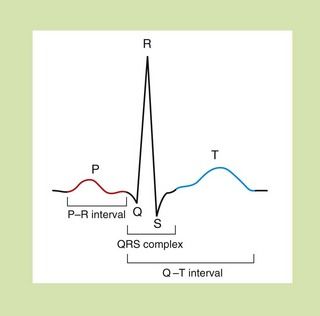
Fig. 8.3 The waveform for cardiac events seen on a surface electrocardiogram. The P wave represents the spread of depolarisation through the atria, and the QRS complex is the spread through the ventricles. The T wave represents repolarisation of the ventricle. The P–R interval is the time of conductance from atrium to ventricles, and the QRS time is the time the ventricles are activated. The duration of the ventricle action potential is given by the Q–T interval.
Mechanisms of arrhythmogenesis
Arrhythmias are disorders of rate and rhythm of the heart, which can arise as the result of either abnormal impulse generation or abnormal impulse conduction. There are three principal mechanisms of arrhythmogenesis.
 Increased automaticity. Dominant ectopic pacemakers (pacemakers other than the SA node) can arise when pacemaker cells in the specialised conducting tissue develop a more rapid phase 4 depolarisation than the SA node. Ectopic pacemakers can also arise when rapid spontaneous phase 4 depolarisation develops in myocytes that usually have a stable phase 4. Ischaemia, or other changes in the microcellular environment, can create conditions that allow a non-specialised myocardial cell to express f-channels and become a pacemaker.
Increased automaticity. Dominant ectopic pacemakers (pacemakers other than the SA node) can arise when pacemaker cells in the specialised conducting tissue develop a more rapid phase 4 depolarisation than the SA node. Ectopic pacemakers can also arise when rapid spontaneous phase 4 depolarisation develops in myocytes that usually have a stable phase 4. Ischaemia, or other changes in the microcellular environment, can create conditions that allow a non-specialised myocardial cell to express f-channels and become a pacemaker. Re-entry. This is the cause of most clinically important arrhythmias. It is initiated when a depolarising impulse (often a premature ectopic beat from elsewhere in the heart) arrives at a part of the myocardium that is still in its refractory period. This is usually a fast conducting pathway with slow recovery from depolarisation. The impulse will bypass the refractory tissue. If this impulse is conducted through an adjacent part of the myocardium with slower conduction but fast recovery, then the impulse may subsequently arrive at the distal part of the refractory tissue when this tissue has had sufficient time to repolarise. The impulse will then be conducted retrogradely through this previously refractory tissue (Fig. 8.4C). If there has been sufficient time for the fast recovery myocardium proximal to the block to repolarise, a self-perpetuating circuit of electrical activity will be initiated (a re-entry circuit). The re-entry circuit acts as a pacemaker that initiates impulses that are then propagated through the heart. Such functional re-entry circuits can be localised within a small area of myocardium that has been damaged by fibrosis or ischaemia (micro re-entry). Micro re-entry circuits may arise in the atria, within the AV node or in the ventricles. The myocardium can also support large anatomical re-entry circuits (macro re-entry). These circuits arise when there is a congenital accessory pathway that bypasses the AV node, and conducts electrical activity between the atria and ventricles. The re-entry circuit between the atria and ventricles includes both the accessory pathway and the AV node (such as occurs in the Wolff–Parkinson–White syndrome).
Re-entry. This is the cause of most clinically important arrhythmias. It is initiated when a depolarising impulse (often a premature ectopic beat from elsewhere in the heart) arrives at a part of the myocardium that is still in its refractory period. This is usually a fast conducting pathway with slow recovery from depolarisation. The impulse will bypass the refractory tissue. If this impulse is conducted through an adjacent part of the myocardium with slower conduction but fast recovery, then the impulse may subsequently arrive at the distal part of the refractory tissue when this tissue has had sufficient time to repolarise. The impulse will then be conducted retrogradely through this previously refractory tissue (Fig. 8.4C). If there has been sufficient time for the fast recovery myocardium proximal to the block to repolarise, a self-perpetuating circuit of electrical activity will be initiated (a re-entry circuit). The re-entry circuit acts as a pacemaker that initiates impulses that are then propagated through the heart. Such functional re-entry circuits can be localised within a small area of myocardium that has been damaged by fibrosis or ischaemia (micro re-entry). Micro re-entry circuits may arise in the atria, within the AV node or in the ventricles. The myocardium can also support large anatomical re-entry circuits (macro re-entry). These circuits arise when there is a congenital accessory pathway that bypasses the AV node, and conducts electrical activity between the atria and ventricles. The re-entry circuit between the atria and ventricles includes both the accessory pathway and the AV node (such as occurs in the Wolff–Parkinson–White syndrome).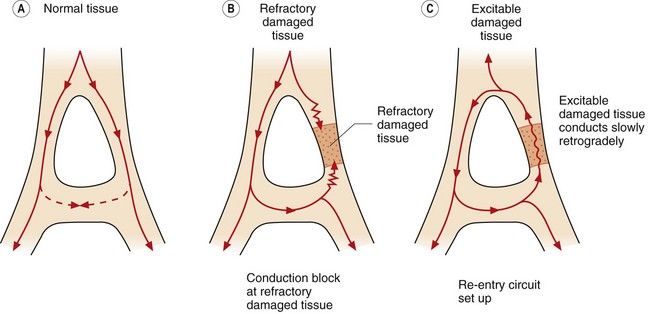
Fig. 8.4 Conduction in normal and damaged cardiac tissue. (A) In normal tissue, conduction is carefully ordered. When an action potential has been generated, the cells cannot be immediately reactivated because of refractoriness of the myocardial cells. If conducted impulses meet, they die out. (B) If an area of damage or dysfunction is present, impulses are conducted abnormally. If an action potential arrives at the area of damaged tissue and it is fully refractory, the impulse is blocked and an arrhythmia will not develop. (C) If an action potential arrives at a damaged area and it is capable of being excited and conducting in a retrograde direction, a perpetuating abnormal re-entry circuit may be set up.
 Triggered activity. A cell can develop transient depolarisations during or following repolarisation (‘afterdepolarisations’), which will initiate an action potential if they reach the threshold potential of the cell. Afterdepolarisations are said to be ‘early’ if they occur during repolarisation (relative refractory period in Fig. 8.1B), or ‘delayed’ if they occur in phase 4. Early afterdepolarisations are due to opening of L-type Ca2+ channels or fast Na+ channels. Delayed afterdepolarisations are due to intracellular Ca2+ overload and follow spontaneous Ca2+ release from the sarcoplasmic reticulum during prolonged repolarisation. This activates the 3Na+/Ca2+ exchange transporter and produces a net depolarising current. Triggered activity is an uncommon mechanism of arrhythmogenesis but may be responsible for the proarrhythmic activity of class Ia and III antiarrhythmic agents (early afterdepolarisations) and digitalis glycosides (delayed afterdepolarisations) (see below).
Triggered activity. A cell can develop transient depolarisations during or following repolarisation (‘afterdepolarisations’), which will initiate an action potential if they reach the threshold potential of the cell. Afterdepolarisations are said to be ‘early’ if they occur during repolarisation (relative refractory period in Fig. 8.1B), or ‘delayed’ if they occur in phase 4. Early afterdepolarisations are due to opening of L-type Ca2+ channels or fast Na+ channels. Delayed afterdepolarisations are due to intracellular Ca2+ overload and follow spontaneous Ca2+ release from the sarcoplasmic reticulum during prolonged repolarisation. This activates the 3Na+/Ca2+ exchange transporter and produces a net depolarising current. Triggered activity is an uncommon mechanism of arrhythmogenesis but may be responsible for the proarrhythmic activity of class Ia and III antiarrhythmic agents (early afterdepolarisations) and digitalis glycosides (delayed afterdepolarisations) (see below).Classification of antiarrhythmic drugs
A widely used classification of antiarrhythmic drugs (the Vaughan Williams classification) is based on their effects on the action potential (Fig. 8.5). This classification has many flaws and does not take account of the multiple actions possessed by some drugs, or the fact that the effects of drugs on diseased myocardium may be different from that on healthy myocardium. However, there is no widely accepted alternative classification.
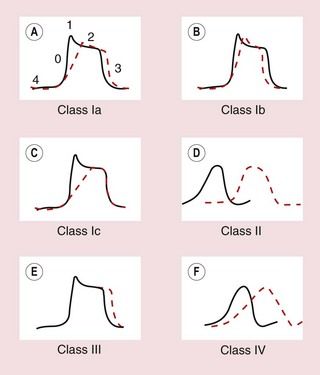
Fig. 8.5 Effects of different classes of antiarrhythmic drug on the cardiac action potential. (A, B, C, E) Drug effects on ventricular cells; (D, F) effects on the AV node. (A) Class Ia drugs. Block fast Na+ channels in phase 0 with moderate potency, and some K+ channels. Repolarisation is prolonged. (B) Class Ib drugs. Weakly block fast Na+ channels in phase 0 only in abnormal tissue; little effect on K+ channels. (C) Class Ic drugs. Potently block fast Na+ channels and weakly block Ca2+ channels and some K+ channels. (D) Class II drugs. Reduce phase 4 and phase 0 depolarisation in AV and SA nodes. Repolarisation in the AV node is prolonged. (E) Class III drugs. Block some K+ channels, inhibiting repolarisation and prolonging the action potential. (F) Class IV drugs. Block L-type Ca2+ channels, slowing phase 0 and phase 4 depolarisation, particularly in the AV node, with less effect in the SA node. Repolarisation is prolonged.
Vaughan Williams classification
The Vaughan Williams classification recognises four classes of drug (Table 8.2).

Class I
All class I drugs inhibit fast Na+ channels and slow the rate of rise of phase 0, and therefore reduce the excitability of the myocardial cell. They are often called membrane stabilisers. They readily penetrate the phospholipid bilayers of the cell membrane, where they concentrate in the hydrophobic core and bind to hydrophobic amino acids in the Na+ channel. Class I drugs are subdivided according to their effects on the duration of the action potential.
 Class Ia drugs, such as disopyramide, slow impulse conduction by producing moderate Na+ channel blockade. In addition, they block some K+ channels (Table 8.1, Figs 8.1 and 8.5A), which prolongs repolarisation and therefore extends the duration of the action potential. They are effective for the treatment of both supraventricular and ventricular arrhythmias (Table 8.2).
Class Ia drugs, such as disopyramide, slow impulse conduction by producing moderate Na+ channel blockade. In addition, they block some K+ channels (Table 8.1, Figs 8.1 and 8.5A), which prolongs repolarisation and therefore extends the duration of the action potential. They are effective for the treatment of both supraventricular and ventricular arrhythmias (Table 8.2). Class Ib drugs, such as lidocaine, slow impulse conduction by producing weak Na+ channel blockade in abnormal tissue (such as ischaemic myocardium) with no effect in healthy tissue. They do not block K+ channels and have either no effect on repolarisation or may shorten it (Fig. 8.5B). They are only effective for the treatment of ventricular arrhythmias.
Class Ib drugs, such as lidocaine, slow impulse conduction by producing weak Na+ channel blockade in abnormal tissue (such as ischaemic myocardium) with no effect in healthy tissue. They do not block K+ channels and have either no effect on repolarisation or may shorten it (Fig. 8.5B). They are only effective for the treatment of ventricular arrhythmias. Class Ic drugs, such as flecainide, slow impulse conduction by producing marked Na+ channel blockade. They produce weak blockade of some K+ channels (Table 8.1), and also block inward Ca2+ channels. There is minimal effect on repolarisation (Fig. 8.5C). They are effective for the treatment of both atrial and ventricular arrhythmias.
Class Ic drugs, such as flecainide, slow impulse conduction by producing marked Na+ channel blockade. They produce weak blockade of some K+ channels (Table 8.1), and also block inward Ca2+ channels. There is minimal effect on repolarisation (Fig. 8.5C). They are effective for the treatment of both atrial and ventricular arrhythmias.The different effects of the class I subgroups result from their diverse ion-channel binding characteristics. During the time course of the action potential, the access of the drug to its binding site is intermittent and dependent on the state of the channel. Class Ib drugs (such as lidocaine) show marked use-dependency; that is, the action increases with the frequency of opening of the channel. They associate more rapidly with Na+ channels during depolarisation and they rapidly dissociate from the channel when it returns to the resting state. Therefore they are more effective when there are repetitive depolarisations, and they will effectively block premature impulses. Cells in ischaemic myocardium are more likely to be depolarised, which explains the selectivity of class Ib drugs for ventricular arrhythmias in ischaemic heart disease (ischaemia mainly affects the ventricles). Class Ic drugs (such as flecainide) also show use-dependent binding, but dissociate slowly from their binding sites in the Na+ channel and therefore produce prolonged blockade. This results in a widespread reduction in cellular excitability. Class Ia drugs (such as disopyramide) have binding characteristics between those of the other two subgroups.
Class II
The class II drugs are the β-adrenoceptor antagonists (β-blockers) which block the actions of catecholamines on the heart. They reduce the rate of spontaneous depolarisation of SA and AV nodal tissue and reduce conduction through the AV node through their action on adrenoceptor-sensitive f-channels (Fig. 8.5D). They can also inhibit ectopic pacemakers that have developed automaticity. Beta-adrenoceptor antagonists are effective for the treatment of both supraventricular and ventricular arrhythmias, particularly if these are catecholamine-dependent.
Class III
Class III drugs such as amiodarone prolong the duration of the action potential by inhibiting some K+ channels involved in repolarisation, thus increasing the absolute refractory period (Table 8.1, Fig. 8.5E). They are effective for the treatment of both supraventricular and ventricular arrhythmias.
Class IV
There are two types of Ca2+ channel in the heart but the class IV drugs are calcium channel blockers that selectively block the L-type Ca2+ channel. They slow conduction of the action potential particularly in the AV node. They have a lesser effect on the rate of depolarisation at the SA node (Fig. 8.5F). They are only effective for the treatment of supraventricular arrhythmias.
Proarrhythmic activity of antiarrhythmic drugs
Many antiarrhythmic drugs have the potential to precipitate serious arrhythmias, such as incessant ventricular tachycardia. Several of them (particularly class Ia agents and sotalol) prolong the Q–T interval on the ECG (Fig. 8.3). This predisposes to a polymorphic ventricular tachycardia known as torsade de pointes, which has a characteristic twisting QRS axis on the ECG and can degenerate into ventricular fibrillation. Drug-induced ventricular rhythm disturbances are particularly refractory to treatment.
There are probably multiple mechanisms of drug-induced arrhythmogenesis. Several risk factors have been identified, including the following.
 Excessive slowing of cardiac impulse conduction, such as occurs with marked blockade of Na+ channels.
Excessive slowing of cardiac impulse conduction, such as occurs with marked blockade of Na+ channels. Excessive prolongation of the action potential, especially if this is due to blockade of the IKr repolarising current (Table 8.1, Fig. 8.2). Prolonged repolarisation may cause early afterdepolarisations (see above), or a selective effect of the drugs on some cells in the myocardium can lead to differential rates of repolarisation that predisposes to re-entry circuits.
Excessive prolongation of the action potential, especially if this is due to blockade of the IKr repolarising current (Table 8.1, Fig. 8.2). Prolonged repolarisation may cause early afterdepolarisations (see above), or a selective effect of the drugs on some cells in the myocardium can lead to differential rates of repolarisation that predisposes to re-entry circuits. Mutations in genes coding for channels that regulate Na+, K+ and Ca2+ transmembrane ion flows exist in 5–10% of people, and are probably subclinical variants of the congenital long QT syndrome. These individuals are more susceptible to torsade de pointes when exposed to a drug that prolongs the Q–T interval.
Mutations in genes coding for channels that regulate Na+, K+ and Ca2+ transmembrane ion flows exist in 5–10% of people, and are probably subclinical variants of the congenital long QT syndrome. These individuals are more susceptible to torsade de pointes when exposed to a drug that prolongs the Q–T interval. Structural heart disease, especially ischaemic heart disease, with greater slowing of conduction in diseased myocardium.
Structural heart disease, especially ischaemic heart disease, with greater slowing of conduction in diseased myocardium.

Class Ia drugs
Disopyramide is no longer widely used in the UK because of its unwanted effects.
Pharmacokinetics: Oral absorption of disopyramide is almost complete. Metabolism in the liver generates a compound with less antiarrhythmic activity but with greater antimuscarinic activity.
< div class='tao-gold-member'>

Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


