Starch, found in plants, is a mixture of amylose (straight chains) and amylopectin (branched chains).
Glycogen, found in animal tissue, is a highly branched polysaccharide.
Table 12.1 Common reducing and non-reducing sugars | |||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
|
synthesize glucose,
store glucose in significant amounts,
metabolize substrates other than glucose and ketones
plasma ketone concentrations are usually very low and ketones are of little importance as an energy source under physiological conditions,
extract enough glucose from the extracellular fluid (ECF) at low concentrations for its metabolic needs, because entry into brain cells is not facilitated by insulin.
even after a carbohydrate meal. Significant glycosuria usually occurs only if the plasma glucose concentration exceeds about 10 mmol/L – the renal threshold.
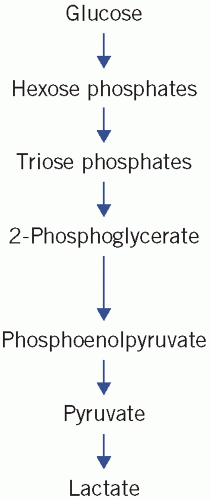 Figure 12.1 Simplification of glycolysis pathways. Reproduced with kind permission from Candlish JK and Crook M. Notes on Clinical Biochemistry. Singapore: World Scientific Publishing, 1993. |
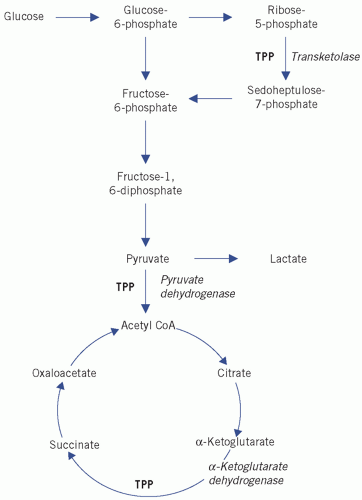 Figure 12.2 Simplification of the tricarboxylic acid (Krebs) cycle. CoA, coenzyme A; TPP, thiamine pyrophosphate. Reproduced with kind permission from Candlish JK and Crook M. Notes on Clinical Biochemistry. Singapore: World Scientific Publishing, 1993. |
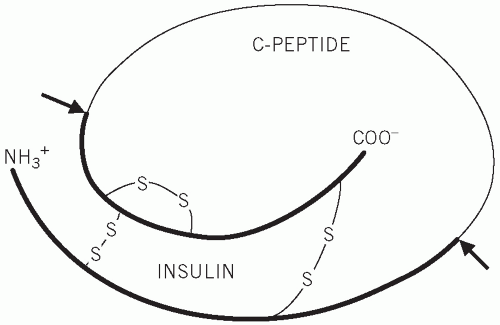 Figure 12.3 Structure of proinsulin, indicating the cleavage sites at which insulin and C-peptide are produced. |
Table 12.2 Action of hormones that affect intermediary metabolism | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
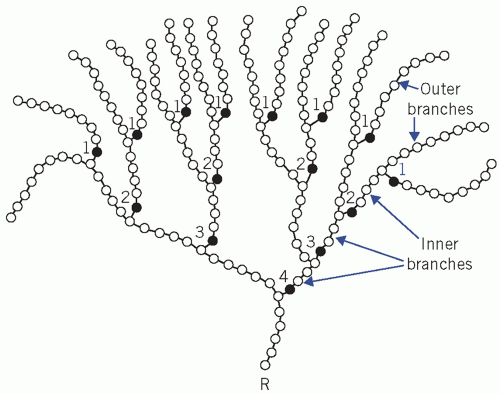 Figure 12.4 Structure of glycogen. Open circles depict glucose moieties in α-1,4 linkage and the black circles those in α-1,6 linkages at branch points. R indicates the reducing end group. The outer branches terminate in non-reducing end groups. Reproduced with permission from Nyhan WL and Barshop BA. Atlas of Inherited Metabolic Diseases, 3rd edition. London: Hodder Arnold, 2012. |
stimulates the β-cells of the pancreas to secrete insulin. Insulin may further enhance hepatic and muscle glycogenesis. More importantly, entry of glucose into adipose tissue and muscle cells, unlike that into liver and brain, is stimulated by insulin and, under physiological conditions, the plasma glucose concentration falls to near fasting levels. Conversion of intracellular glucose to G6P in adipose and muscle cells is catalysed by the enzyme hexokinase, which, because its affinity for glucose is greater than that of hepatic glucokinase, ensures that glucose enters the metabolic pathways in these tissues at lower extracellular concentrations than those in the liver. The relatively high insulin activity after a meal also inhibits the breakdown of triglyceride (lipolysis) and protein (proteolysis). If there is relative or absolute insulin deficiency, as in diabetes mellitus, these actions are impaired. Both muscle and adipose tissue store the excess post-prandial glucose, but the mode of storage and the function of the two types of cell are very different, as will be shown later.
Table 12.3 Metabolism of the carbon skeleton of some amino acids to either carbohydrate (glycogenic) or fat (ketogenic) | ||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
|
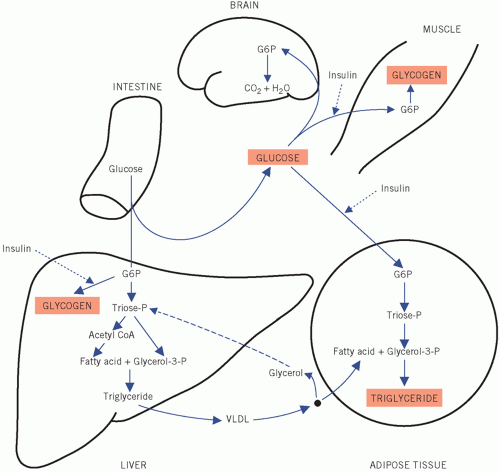
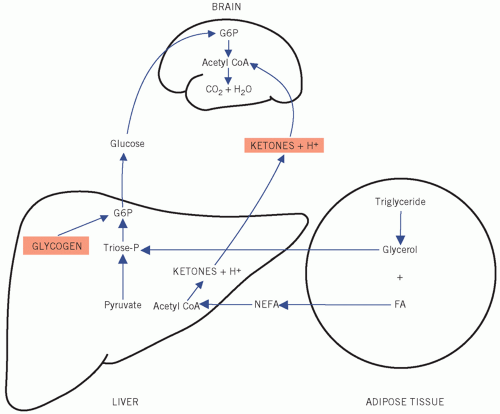 |
??
During gluconeogenesis, hydrogen ions are reused. Under aerobic conditions, the liver consumes much more lactate than it produces.
production is increased by an increased rate of anaerobic glycolysis,
use is decreased by impairment of the TCA cycle or impairment of gluconeogenesis.
the TCA cycle cannot function anaerobically and oxidation of pyruvate and lactate to CO2 and water is impaired,
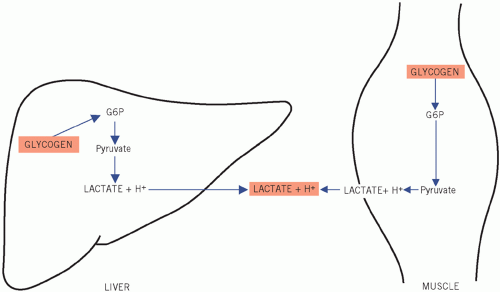
Figure 12.8 Metabolic pathways during 0tissue hypoxia. G6P, glucose-6-phosphate.
hepatic and renal gluconeogenesis from lactate cannot occur anaerobically,
intravenous infusion of glucose-containing fluids,
severe stress (usually a transient effect) such as trauma, myocardial infarction or cerebrovascular accidents,
diabetes mellitus or impaired glucose regulation.
Maturity-onset diabetes of the young (MODY):
Type A insulin resistance (insulin receptor defect), for example leprechaunism, lipoatrophy and Rabson-Mendenhall syndrome.
Chronic pancreatitis.
Pancreatectomy.
Haemochromatosis.
Cystic fibrosis.



