Biotin
Substrate Reduction
As illustrated by the damaging effects of hyperphenylalaninemia in PKU, enzyme deficiencies may lead to substrate accumulation, with pathophysiological consequences (see Chapter 12). Strategies to prevent the accumulation of the offending substrate have been one of the most effective methods of treating genetic disease. The most common approach is to reduce the dietary intake of the substrate or of a precursor of it, and presently several dozen disorders—most involving amino acid catabolic pathways—are managed in this way. The drawback is that severe lifelong restriction of dietary protein intake is often necessary, requiring strict adherence to an artificial diet that is onerous for the family as well as for the patient. Nutrients such as 20 essential amino acids cannot be withheld entirely, however; their intake must be sufficient for anabolic needs such as protein synthesis.
A diet restricted in phenylalanine largely circumvents the neurological damage in classic PKU (see Chapter 12). Phenylketonuric children are normal at birth because the maternal enzyme protects them during prenatal life. Treatment is most effective if begun promptly after diagnosis by newborn screening. Without treatment, irreversible developmental delay occurs, the degree of intellectual deficit being directly related to the delay in commencing the low-phenylalanine diet. It is now recommended that patients with PKU remain on a low-phenylalanine diet for life because neurological and behavioral abnormalities develop in many (although perhaps not all) patients if the diet is stopped. However, even PKU patients who have been effectively treated throughout life may have neuropsychological deficits (e.g., impaired conceptual, visual-spatial, and language skills), despite their having normal intelligence as measured by IQ tests. Nonetheless, treatment produces results vastly superior to the severe developmental delay that occurs without treatment. As discussed in Chapter 12, continued phenylalanine restriction is particularly important in women with PKU during pregnancy to prevent prenatal damage to the fetus, even though the fetus is highly unlikely to be affected by PKU.
Replacement
The provision of essential metabolites, cofactors, or hormones whose deficiency is due to a genetic disease is simple in concept and often simple in application. Some of the most successfully treated single-gene defects belong to this category. A prime example is provided by congenital hypothyroidism, of which 10% to 15% of cases are monogenic in origin. Monogenic congenital hypothyroidism can result from mutations in any one of numerous genes encoding proteins required for the development of the thyroid gland or the biosynthesis or metabolism of thyroxine. Because congenital hypothyroidism from all causes is common (approximately 1 in 4000 neonates), neonatal screening is conducted in many countries so that thyroxine administration may be initiated soon after birth to prevent the severe intellectual defects that are otherwise inevitable (see Chapter 18).
Diversion
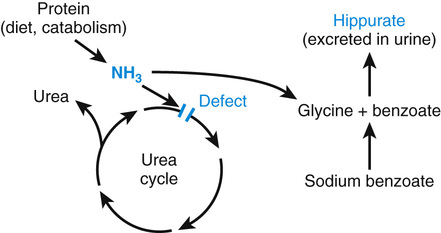
Diversion therapy is the enhanced use of alternative metabolic pathways to reduce the concentration of a harmful metabolite. A major use of this strategy is in the treatment of the urea cycle disorders (Fig. 13-4). The function of the urea cycle is to convert ammonia, which is neurotoxic, to urea, a benign end product of protein catabolism excreted in urine. If the cycle is disrupted by an enzyme defect such as ornithine transcarbamylase deficiency (Case 36), the consequent hyperammonemia can be only partially controlled by dietary protein restriction. Blood ammonia levels can be reduced to normal, however, by the diversion of ammonia to metabolic pathways that are normally of minor significance, leading to the synthesis of harmless compounds. Thus, the administration to hyperammonemic patients of large quantities of sodium benzoate forces the ligation of ammonia with glycine to form hippurate, which is excreted in urine (see Fig. 13-4). Glycine synthesis is thereby increased, and for each mole of glycine formed, one mole of ammonia is consumed.
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


