(1)
Department of Biomedical Engineering, University of Minnesota, Minneapolis, MN, USA
(2)
School of Physics and Astronomy, University of Minnesota, Minneapolis, MN, USA
(3)
Department of Biomedical Engineering, School of Physics and Astronomy, University of Minnesota, Minneapolis, MN, USA
Abstract
This chapter presents an overview of quantitative fluorescence brightness experiments with special emphasis on single-color measurements of protein homo-interactions inside living cells. We discuss practical considerations in the choice of the fluorescent labels and the calibration measurements necessary for quantitative interpretation of brightness experiments. Methods to identify and avoid common pitfalls, such as bleaching and saturation, are addressed. We examine the interpretation of brightness data with moment analysis. In particular, we focus on how to avoid or correct for undersampling, as well as how to characterize and adjust for photon detector effects. We conclude by describing brightness titration experiments which determine the binding curve and stoichiometry of a protein from apparent brightness data.
Key words
StoichiometryFluctuationTitrationTwo-photonPhoton count momentsAutocorrelationDead timeAfterpulsingPhotobleaching1 Introduction
Fluorescently tagged biomolecules passing through a small optical observation volume give rise to fluorescence bursts. The frequency, duration, and amplitude of these bursts are the main sources of information for most fluorescence fluctuation spectroscopy (FFS) experiments. Here we focus on brightness, which is closely connected to the amplitude of the fluorescence fluctuations [1]. Brightness represents the average photon count rate emitted by a fluorescent bioparticle and is typically expressed in units of counts per second (cps). The main application discussed in this chapter is the calculation of the average number of fluorescently tagged biomolecules per molecular complex. Thus, brightness analysis provides information about the average stoichiometry of a sample. While brightness experiments can be performed both in cells and solution, we emphasize cellular applications, which are the most challenging and most interesting applications of brightness analysis.
Our group focuses on two-photon excitation measurements, which are reflected in this chapter. Nevertheless, the selection of fluorophores, measurement of cells, and analysis of data present challenges that are largely independent of the excitation method. We discuss common pitfalls and suggest strategies to avoid them. The potential topics relevant to brightness experiments are vast and occasionally intricate. To keep the subject focused and the material manageable, we restrict our discussion to single-color FFS experiments, analysis of photon count moments, and brightness titration. In our experience, single-color brightness titration provides the most straightforward brightness method to quantify protein homo-interactions in cells.
2 Methods
2.1 The Fluorescent Label
The fluorescent label is the fundamental tool of brightness experiments, and great care should be taken in its selection and characterization. In this chapter we limit ourselves to genetic tagging, i.e., the use of fluorescent proteins as labels. The number of fluorescent proteins available is now quite large and continues to grow, but not all are suitable for brightness measurements. Brightness is important because it encodes stoichiometry. To emphasize this point and to simplify discussion, we introduce the concept of dimensionless brightness (also referred to as normalized brightness),
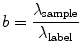
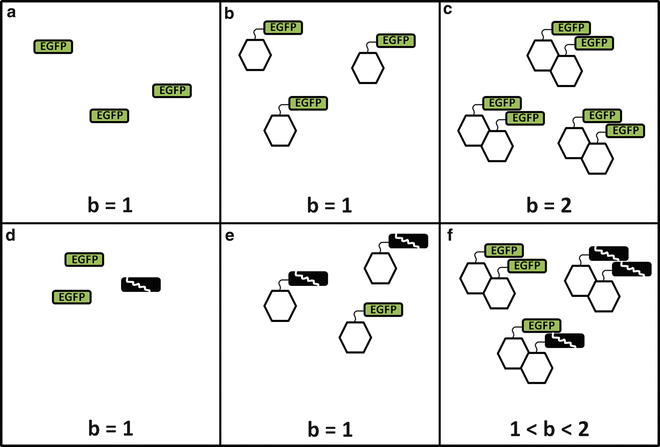
where λ sample is the brightness of the sample and λ label is the brightness of the label alone (Fig. 1a). By this definition, b = 1 represents a monomeric sample (Fig. 1b), and b = 2 is a dimeric sample (Fig. 1c). In later sections, we will critically evaluate this straightforward relationship between brightness and protein stoichiometry and generalize it.
(1)
Fig. 1
Brightness, labels, and stoichiometry. The top rank indicates a conceptual picture for the fluorophore EGFP with a single brightness state. Once the inherent brightness of the fluorescent label (a) is identified, the information can be used to identify the oligomeric state of a protein of interest (depicted as a hexagon). If the protein is monomeric (b), the brightness returns a normalized value of 1, while if the protein is dimeric (c), the normalized brightness equals 2. However, if a fraction of the label is in a nonfluorescent state, neither the label calibration (d) nor a monomeric protein (e) will detect the presence of the dark label. In contrast, for a dimeric protein (f), the presence of dark label induces a mixture of brightness species, which returns an apparent brightness that falls between the monomer and the dimer
2.1.1 Fluorophore Brightness Properties
Because the determination of stoichiometry is predicated on one label per protein, the selected fluorophore label must exist as a monomer in its natural state. Fluorescence proteins that self-associate to form dimers or tetramers ought not to be used for brightness experiments because the measured interactions stem, at least partially, from the labels rather than from the proteins of interest. Therefore these labels are not faithful reporters of the interactions of the protein of interest.
One also needs to be aware of additional properties of the fluorescent label. Ideally, a fluorescent protein exhibits a single brightness state. In this case, the normalized brightness directly reflects the oligomeric state of the protein, i.e., an n-mer labeled protein complex has a brightness b = n. Enhanced green fluorescent protein (EGFP) and enhanced yellow fluorescent protein (EYFP) each have, for all practical purposes, only one brightness state. Unfortunately, some fluorescent proteins exist in more than a single brightness state. We first discuss the special case where a fraction of the labels is nonfluorescent. The nonfluorescent state of a label is also referred to as a dark state. The presence of labels in the dark state destroys straightforward interpretation of the normalized brightness and can lead to misinterpretation of the data. For example, consider a dimeric sample with fluorophore labels, some fraction of which is in a dark state (Fig. 1d). In a monomer sample the brightness will be unaffected, as only the fluorophores which are “on” will be detected (Fig. 1e). However, in a dimer sample, a mixture of bright and dark labels will lead to three populations: bright-bright, dark-dark, and dark-bright (Fig. 1f). The first species returns the expected normalized brightness of b = 2, the second species is nonfluorescent and thus not detected, and the third species returns b = 1. An FFS experiment of such a mixture usually identifies only a single brightness (see Note 1), which we refer to as apparent brightness 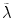 . Its value represents the ensemble average over all brightness species (see Note 2). The specific value depends on the fractional populations of the three species depicted in Fig. 1f, but it is clear that the apparent brightness for this case will fall between 1 and 2 [2].
. Its value represents the ensemble average over all brightness species (see Note 2). The specific value depends on the fractional populations of the three species depicted in Fig. 1f, but it is clear that the apparent brightness for this case will fall between 1 and 2 [2].
. Its value represents the ensemble average over all brightness species (see Note 2). The specific value depends on the fractional populations of the three species depicted in Fig. 1f, but it is clear that the apparent brightness for this case will fall between 1 and 2 [2].Because dark states complicate the interpretation of brightness experiments, it is best to avoid the various conditions that introduce dark states. As an example, photobleaching of the sample introduces a population of labels in the dark state. Similarly, the slow maturation kinetics of some fluorescent proteins causes a significant delay between the folding of the protein and the onset of fluorescence, which generates a population of proteins with nonfluorescent labels. Thus, it is prudent to avoid slowly maturing fluorescent proteins, experimental conditions that lead to photobleaching, and fluorescent proteins with an intrinsic population of dark states (see Note 3).
The presence of a bright and a dark state is just a special case of brightness heterogeneity of the label (see Note 4). The red fluorescent protein mCherry is another example of brightness heterogeneity and has been successfully modeled as a mixture of a long-lived bright and dim state [3]. Consequently, an mCherry-labeled dimer consists of a mixture of three brightness species (bright-bright, dim-dim, and dim-bright), which leads to a normalized brightness of less than 2. In general, brightness heterogeneity should be avoided, because it also destroys the simple relationship between stoichiometry and normalized brightness. However, brightness heterogeneity of the label does not necessarily prohibit its use in brightness experiments as long as the brightness states of the label are well characterized [3]. We provide a concrete example in Subheading 2.2.
Finally, some fluorophores are brighter than others at an equivalent excitation power. It is difficult to get good signal statistics from dim proteins especially in cells which have non-negligible autofluorescence. Enhancing brightness by increasing the excitation power is limited by the onset of photobleaching. Thus, the selection of the label should also take the intrinsic brightness of the fluorescent protein into account.
2.1.2 Photobleaching and Saturation
Photobleaching is the permanent loss of fluorescence in a fluorophore caused by photon-induced chemical damage. The literature [4–6] rates fluorescent proteins based on photostability and one must choose the most stable fluorophore that is suitable for the proposed experiment. It is also necessary to consider the saturation level of the fluorophore. A fluorophore in the excited state has a finite lifetime before emitting a photon and relaxing back to the ground state. Because of this, there is a maximum rate beyond which the fluorophore will not respond to changes in the excitation intensity as expected. Brightness experiments should stay well below this regime.
To characterize a fluorophore with regard to these phenomena, we begin by performing a power study, measuring the fluorophore at a series of excitation powers and recording the results. For two-photon excitation, both the fluorescence intensity and brightness should scale with the square of the excitation power, but brightness is the more sensitive parameter (see Note 5). A plot of the measured brightness as a function of power squared (for 2-photon excitation) visually identifies the power at which the brightness is starting to be sublinear. We choose a point approximately 10 % below that divergence as the maximum allowable excitation power. Photobleaching may be detected through a decrease in the fluorescence intensity during the course of the measurement because the cumulative damage leads to a reduction of the fluorescent label population within the finite volume of the cell. The autocorrelation function can also detect photobleaching through a reduction in diffusion time, but this only becomes apparent for severe photobleaching. We prefer the much more sensitive approach of monitoring the fluorescence intensity over a long data acquisition. Regardless, detectable levels of photobleaching are clearly detrimental to brightness measurements as they create a population of nonfluorescent labels and reduce the normalized brightness in the same fashion as was discussed for long-lived dark states. Therefore, one should select a photostable fluorophore and choose conditions that ensure negligible photobleaching.
On the whole, the fluorophore is to a fluorescence experimentalist as is the voice to an opera singer. If one is not aware of and comfortable with all its nuances, strengths, and flaws, the result will not be worth the price of the ticket.
2.2 Calibrations
Another critical feature of a quantitative brightness measurement is a careful calibration. By its nature, the fluctuation intensity signal is noisy and provides little in the way of observable phenomena or immediate evidence of underlying problems. Furthermore, the power of brightness analysis to determine stoichiometry is directly dependent on the brightness of the protein complex relative to the brightness of the chosen label. Therefore, both the successful application of the technique and the careful interpretation of inherently noisy data are founded upon control experiments. Calibrations are required on two levels. The first is a daily measurement of a well-known dye sample. This serves as a standard for the long-term performance of the instrument and an alert to any problems with the laser, the instrument, or the alignment thereof. One common instrument issue is a straightforward—but undocumented—alteration by the previous user. In addition, small particles of dust or finger prints on the optics, slight relaxations of a mirror or dichroic filter, and—in one notable instance—a spider web in the laser cavity have each caused a deterioration in instrument performance that we might not have easily or quickly detected without the record of a daily instrument calibration. Daily brightness (see Note 5) calibrations prevent such issues from spoiling an experiment and provide a baseline for further optimization. In our calibration procedure, we prepare a quantity of a bright dye solution and then aliquot and freeze it, so that each day’s experiment begins with a control measurement performed in the same way, on the same sample, and at the same distance focused into the sample (see Note 6). In this way, we track the operation of the instrument not only from day to day but from year to year.
The second level of calibration is the sample calibration that establishes the baseline brightness for a given sample preparation. The basic sample calibrations for any brightness experiment in cells involve measuring both the monomeric label and a dimeric construct of said label (generated by cloning a second EGFP sequence into a vector alongside the original EGFP sequence) [2]. These measurements provide controls to relate brightness to stoichiometry. At the simplest level, the monomer sample provides a simple reference point for normalized brightness, and the dimer sample indicates that the brightness measurement and analysis are proceeding as expected, namely,
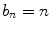
where b n is the normalized brightness of an n-mer sample. Under further consideration, there is more to be learned. Both calibrations should cover a broad range of concentrations by selecting cells that have high, medium, and low expression levels of the protein. A good label will have a constant brightness independent of concentration, demonstrating that brightness is a robust value and that the label will not induce interactions on its own. A successful dimer calibration should return a normalized brightness very close to 2. This indicates the absence of significant brightness heterogeneity of the label, such as introduced by photobleaching, unmatured fluorophores, or dark states. Table 1 shows monomer/dimer label calibrations in different cell lines for the three fluorophores most commonly used in our lab. EGFP and EYFP show good doubling within measurement error, indicating the presence of a single brightness state. However, mCherry has a value less than 2, revealing the presence of brightness heterogeneity of the label. In this case, the expected brightness of an n-mer sample can still be determined from a good monomer/dimer label calibration:
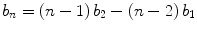
where b 2 is the experimentally determined brightness from the dimeric label calibration and b 1 ≡ 1. (Note that Eq. 3 reduces to Eq. 2 when b 2 = 2.) It is always best to choose a fluorophore with a single, uniform brightness state and thus have the simple brightness-stoichiometry relationship. However, when circumstances (like color choice) drive the fluorophore selection, proteins with brightness heterogeneity, like mCherry, can still be made to work in quantitative brightness experiments, provided that those fluorophores are well characterized. Brightness experiments can quickly and simply provide stoichiometric information, but the experiments must be well designed and carefully interpreted. Good calibrations are at the core of this methodology (see Note 7).
(2)
Table 1
Fluorescent protein dimer calibrations
Normalized brightness, b | EYFP2 | EGFP2 | mCherry2 |
|---|---|---|---|
293T cells | 1.95 ± 0.06 | 1.90 ± 0.07 | 1.62 ± 0.12 |
COS-1 cells | 1.92 ± 0.07 | 1.95 ± 0.05 | 1.65 ± 0.10 |
(3)
2.3 Cell Measurements
The first practical consideration in cell measurements is the choice of cell line. In most cases, this will be driven by the relevant biology, but it is worth mentioning that choosing cells that are easy to transfect, adhere well to surfaces, and are large in size simplifies brightness measurements. Having chosen a cell line, it is important to become familiar with it. Cells expressing the labeled protein of interest should look just the same as untransfected cells. Otherwise, a cell under stress may exhibit abnormal biology, adding scatter or a bias to the brightness data. We consider it good practice to make a brief inspection of every cell before measurement to ensure that the morphology appears healthy.
The next goal is to avoid photobleaching of the labeled proteins. Not all of the prepared cells express the labeled protein, so efficient identification and selection of measureable cells requires viewing the cells under epifluorescent light. This also makes it possible to more evenly sample across a broad protein concentration range. However, it is very important to limit such wide-field excitation of the cells to preclude photobleaching, so we endeavor to make our cell selections “briskly,” only turning on the lamp when actively viewing the cells. Before beginning the experiment, select a cell and illuminate it under epifluorescence for a long period while monitoring the fluorescence intensity. Having identified the characteristic time for decay of the fluorescence signal, one can select a shorter period during which epifluorescent photobleaching is negligible. Similarly, it is vital to limit the photobleaching caused by the laser excitation while measuring. Continuous decay in the fluorescence intensity during the measurement points toward a reduction of the fluorescent population due to photobleaching. We recommend taking some measurements 5× longer than the typical acquisition. If there is no decay in the fluorescence trace, photobleaching effects may be considered negligible at that power for that fluorophore (see Note 8). If photobleaching is detected, reduce the excitation power. However, reducing the excitation power also produces a weaker fluctuation signal which imposes a lower limit on this approach. In the event that one cannot identify an excitation power that has a sufficiently strong signal and avoids photobleaching, it is necessary to redesign the experiment.
Lastly, when planning a cell measurement, it is important to consider the geometry of the cell and protein localization within it. For a nuclear protein, we position the excitation volume in the nucleus, which, aside from the easily seen nucleoli, is an open space free from organelles and internal membranes. Importantly, the nucleus is typically large enough to completely contain the excitation volume as depicted in Fig. 2a. In this scenario, there is usually little cause for concern (see Note 9). However, if the protein must be measured in the cytoplasm, the height of the cell will most likely be shorter than the excitation volume. In this case (Fig. 2b), the proteins are restricted to the central, high-intensity region of the excitation volume which will bias the calculated brightness upwards. Using a z-scan approach can identify and correct for such thickness-dependent artifacts [7].
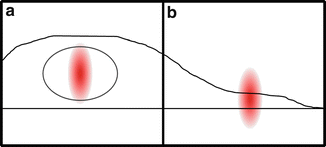
Fig. 2




Cell geometry. The relationship between the PSF of the laser excitation and cell geometry plays an important role in the interpretation of quantitative brightness experiments. When measuring in the nuclei of most cell lines, the PSF will be completely contained (a) and the sample can be considered effectively infinite in thickness. (b) In thin samples, like cell cytoplasm, the PSF will likely extend beyond the sample, limiting the region of excitation accessible to the fluorophores. In such a case, or when membrane-bound protein fractions create inhomogeneous layers, a z-scan approach and theory must be used to recover accurate data
Stay updated, free articles. Join our Telegram channel
Full access? Get Clinical Tree
