Fig. 17.1.
Chronic myelogenous leukemia. Peripheral blood smear with increased granulocytes with left shift, including increased basophils.
Definition
♦
MPN associated with the BCR/ABL1 gene fusion of marrow stem cells, leading most often to blood and marrow expansion of predominantly granulocytic elements
Clinical
♦
Clinical presentation: fatigue (anemia), fullness/LUQ pain (splenomegaly), fever, and malaise are common findings
♦
Chronic myelogenous leukemia (CML) is a disease of adults, most often in their sixth decade. CML very rarely occurs in children but should be distinguished from juvenile myelomonocytic leukemia (see below)
Diagnosis
♦
Peripheral blood
Leukocytosis, often >100 × 109/L, composed of neutrophils and immature forms (predominantly neutrophilic) but <10% blasts. Basophilia is typical, and eosinophilia may also be prominent. Immature erythroid elements (nucleated red blood cells) and thrombocytosis are variably present
♦
Aspirate smear
Hypercellular smears consisting of numerous maturing granulocytic elements are characteristic. The M:E ratio is usually greater than 20:1. Dysplasia of the myeloid series is not typically present. Blasts number <10% in the chronic phase of CML. Megakaryocytes often increased and many have a distinctive morphology; these so-called dwarf forms have small hypolobated nuclei and small amounts of mature cytoplasm. Pseudo-Gaucher cells (macrophages with sea-blue cytoplasm) may be seen
♦
Biopsy
Cores are hypercellular (often approaching 100%), and composed primarily of myeloid precursors. Often a paratrabecular expansion of immature myeloid elements is seen, causing a “myeloid bulge.” This is an increase in the amount of immature myeloid elements seen at the bone, an extending into the intertrabecular space. Dwarf (small, hypolobated) megakaryocytes are common. Marrow sinuses are often dilated and frequently have intrasinusoidal hematopoiesis
Additional Studies
♦
Flow: Although flow cytometry is not uniformly used, abnormalities of myeloid antigen expression and maturation (e.g., myeloid expression of CD56) can be seen. These findings tend to occur in all cases but are not entirely specific for CML
♦
IHC: Rarely used. CD34 staining may highlight increased blasts (see accelerated phase, below)
Cytogenetics/Molecular
♦
Characteristic translocation between chromosomes 9 and 22, leading to the formation of Philadelphia chromosome (Ph1). The t(9;22)(q34;q11) juxtaposes the BCR and ABL1 genes leading to an overactive tyrosine kinase
♦
Although most often the translocation can be identified by routine karyotype, a small subset of cases have cryptic translocations not seen by conventional cytogenetic analysis. Thus, FISH studies are the most sensitive way to detect the t(9;22) at diagnosis
♦
p210 gene product: most common (95%) – typical CML
♦
p190 gene product: rare (~5%) – morphology similar to CMML (also seen in ALL )
♦
p230 gene product: very rare (<1%) – morphology essential thrombocythemia (ET)-like or very rarely CNL-like
Differential Diagnosis
♦
Leukemoid reaction: reactive increase of granulocytes, without cytogenetic change
♦
Chronic neutrophilic leukemia (see below), chronic eosinophilic leukemia (see below), and atypical CML (lacks cytogenetic and molecular evidence of BCR/ABL1 translocation)
Treatment and Prognosis
♦
Family of tyrosine kinase inhibitors (e.g., imatinib) has proven to be very effective in the treatment of CML
♦
Effectiveness of therapy often followed by quantitative RT-PCR studies of BCR/ABL1 expression in peripheral blood
♦
BMT may be the therapy of choice in some patients
CML: Accelerated Phase/Blast Phase
Definition
♦
Aggressive forms of CML, with associated worse prognosis:
Accelerated phase (AP): advancement of clinical and laboratory features associated with a worsening prognosis
Blast phase (blast crisis): Clinical characteristics similar to poor prognosis acute leukemia, with >20% blasts in the blood or bone marrow; approximately 30% of blast phase CML are lymphoblastic leukemias
Clinical
♦
Features of accelerated phase include any of the following: (1) 10–19% blasts in the blood or marrow, (2) blood basophils >20%, (3) persistent thrombocytopenia or thrombocytosis in spite of therapy, (4) increasing spleen size and WBC count in spite of therapy, and (5) cytogenetic evidence of clonal evolution
♦
Criteria for blast phase include any of the following: (1) >20% blasts in the blood or marrow, (2) extramedullary blast proliferation (e.g., spleen), and (3) large clusters or aggregates of blasts in the bone marrow (often identified by immunohistochemistry)
Diagnosis
♦
Peripheral blood
As in the criteria above, there are increased blasts (10–19%) or increased basophils (>20%). The presence of dysplastic features in granulocytic elements is a sign of CML-AP
♦
Aspirate smear
The aspirate smears show similar features to peripheral blood including increased blasts (>10%) and dysplastic features of marrow elements
♦
Biopsy
Clusters and aggregates of increased blasts are seen. If present, significant reticulin fibrosis may suggest accelerated phase. Increased numbers of megakaryocytes or large clusters of megakaryocytes may also indicate CML-AP
Additional Studies
♦
Flow: Myeloid blasts (CD13, CD33, CD34, CD117) or lymphoid blasts (CD19, CD10, TdT) may be seen. Often mixed lineage leukemias with combination of myeloid and lymphoid antigens may be seen
♦
IHC: CD34/CD117 may highlight clusters of blasts
♦
Cytogenetics: In addition to t(9;22), additional abnormalities are often present. Most common findings include double Philadelphia chromosome, iso(17q), and trisomy 8
Polycythemia Vera
Definition
♦
Clonal MPN with predominant erythroid proliferation
Clinical
♦
The M:F ratio is 1–2:1
♦
Diagnostic criteria ( both major criteria and one minor criterion or one major criterion and two minor criteria)
♦
Major criteria:
Hemoglobin >18.5 g/dL (men) or 16.5 g/dL (women) or other evidences of increased red blood cell mass
The presence of JAK2 mutation or other functionally similar mutation
♦
Minor criteria:
Hypercellular bone marrow with panmyelosis
Low serum erythropoietin
In vitro endogenous erythroid colony formation
♦
Other clinical findings that are common but do not count as specific criteria are splenomegaly, thrombocytosis >400 × 109/L and WBC >12 × 109/L. Thrombotic symptoms are relatively common
♦
Postpolycythemic myelofibrosis (post-PV-MF) is characterized by marrow fibrosis, splenic enlargement, and leukoerythroblastic peripheral blood. It is considered to be a late stage of the disease. Splenomegaly with marked neoplastic extramedullary myeloid proliferations is common
Diagnosis
♦
Peripheral blood
Leukocytosis and thrombocytosis are common. Basophilia may be present. Red cell abnormalities are usually not striking, although microcytosis is frequently seen
♦
Aspirate smear
Increase in all marrow elements (panmyelosis) lacking dysplasia. Stainable iron is typically absent
♦
Biopsy
Normo- to hypercellular; if hypercellular, typically has increased erythroid elements. Megakaryocytes may be increased and have dysplastic appearance. Post-PV-MF is a late development in PV with extensive fibrosis that has a morphologic appearance that may be indistinguishable from primary myelofibrosis (see below)
Additional Studies
♦
Cytogenetics/molecular: JAK2 mutations (especially JAK2 V617F) are seen in almost all cases of PV. Recurring cytogenetic abnormalities are also seen in many cases. Common cytogenetic changes include trisomy of chromosomes 8 and 9 and deletions of 20q, 13q, and 9p
Differential Diagnosis
♦
Reactive erythrocytosis, other MPNs
Primary Myelofibrosis (Fig. 17.2)
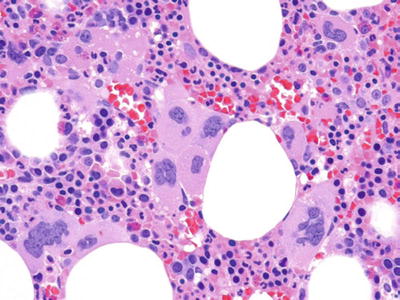
Fig. 17.2.
Primary myelofibrosis. Note the clusters of morphologically atypical megakaryocytes. Extensive fibrosis would be seen on reticulin staining (not shown).
Definition
♦
Clonal MPN characterized by the proliferation of megakaryocytes with significant associated marrow fibrosis
Clinical
♦
Splenomegaly and hepatomegaly due to extramedullary p roliferation of myeloid elements. “Prefibrotic stage” lacks splenomegaly and characteristic peripheral blood findings, lacks marrow fibrosis, but has hypercellularity and megakaryocyte morphologic abnormalities. It accounts for up to 30% of cases of primary myelofibrosis (PMF )
♦
In PMF, transformation to acute myeloid leukemia (AML) occurs in 5–30% of patients
Diagnosis
♦
Peripheral blood
Findings in peripheral blood include leukoerythroblastosis (immature myeloid and erythroid elements), teardrop-shaped red blood cells (dacrocytes), immature neutrophilic elements (including rare blasts), and increased platelets (including abnormal hypogranulated or giant platelets). Rarely, circulating megakaryocytes or megakaryoblasts can be seen
♦
Aspirate smear
Most cases inaspirable due to marrow fibrosis
♦
Biopsy
Increased abnormal megakaryocytes are seen, usually in tight clusters with enlarged, hyperchromatic nuclei. Marrow sinuses are dilated. Intrasinusoidal hematopoiesis is commonly seen within the sinuses. Blasts account for <10% of the marrow cellularity. Reticulin fibrosis is typically present and moderate or severe; cases often have trichrome (mature collagen) positive fibrosis. Bone changes including osteosclerosis and abnormal trabeculae are common findings
Additional Studies
♦
Flow: Flow examination may not be informative due to lack of aspirate
♦
IHC: CD34 may highlight increased blasts and also highlights intravascular hematopoiesis
♦
Cytogenetics/molecular: Approximately 50% of PMF patients will have JAK2 mutation or, less commonly, mutations of MPL. Recently, mutations in calreticulin (CALR) have been identified in the majority of cases without the JAK2 or MPL mutation. Cytogenetic abnormalities occur in up to 30% of patients with the most common being del(13q), del(20q), der(6)t(1;6), partial trisomy (1q), trisomy 8, or trisomy 9
Differential Diagnosis
♦
Acute panmyelosis with myelofibrosis, a rare subset of acute myeloid leukemia
♦
Polycythemia vera/post-PV-MF
♦
AML or MDS with fibrosis
♦
Autoimmune myelofibrosis: seen typically in young women with other autoimmune manifestations; lacks megakaryocytic abnormalities; lacks molecular/genetic abnormalities
♦
Prefibrotic phase can clinically mimic essential thrombocythemia, and bone marrow evaluation may be necessary to distinguish these entities
Treatment and Prognosis
♦
Median survival of 3–7 years from diagnosis
Essential Thrombocythemia (Fig. 17.3)
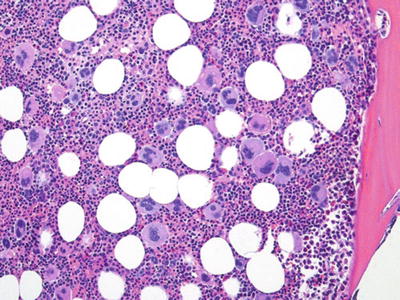
Fig. 17.3.
Essential thrombocythemia. Note increased megakaryocytes. In contrast to PMF, fibrosis is rare.
Definition
♦
A neoplastic proliferation of marrow elements, with a from diagnosispredominance of megakaryocytes; these abnormal megakaryocytes produce large numbers of circulating platelets
Clinical
♦
Patients are often asymptomatic (>50%) or present with consequences of thrombocytosis; these can include either bleeding (hypofunctional platelets) or thrombosis. In contrast to other MPNs, transformation to AML is rare
Diagnosis
♦
Peripheral blood
A sustained platelet count >450 × 109/L is a diagnostic criterion. Platelets are typically morphologically abnormal including variation in size, shape, and granulation. Elevated white blood cell count or basophilia is uncommon
♦
Aspirate smear
Increased megakaryocytes are seen and are typically large in size with large nuclei and abundant cytoplasm. Myeloid and erythroid elements are typically normal in appearance and maturation. Large clumps of platelets may occasionally be seen in aspirate smear
♦
Biopsy
Often normocellular, there is a notable increase in number of megakaryocytes, which are present in loose clusters. The dysplastic megakaryocyte morphology includes very large size, with “cloud-like” nuclei and large amounts of mature, granular cytoplasm. Myeloid and erythroid elements are normal appearing. Reticulin fibrosis is absent or minimal in extent
Additional Studies
♦
Flow/IHC: Occasional dysmaturation of myeloid elements can be seen. However, the findings are not specific
♦
Cytogenetics/molecular: Mutated JAK2 is seen in 40–50% of cases, and MPL mutations are identified less frequently. Similar to primary myelofibrosis, CALR mutations are present in the majority of cases that lack the JAK2 or MPL mutation. Abnormal cytogenetics are rare but include del(20q) trisomy 8 and abnormalities of 9q in a small percentage of cases
Differential Diagnosis
♦
Reactive thrombocytosis lacks morphologic abnormalities in megakaryocytes. Also there are no abnormal cytogenetic or molecular findings
♦
Lacks features of other MPNs (including CML, PMF, post-PV-MF, PV) and MDS
Treatment and Prognosis
♦
Indolent disorder; transformation to AML is rare (<5%)
Myeloid and Lymphoid Neoplasms with Eosinophilia and Abnormalities of PDGFRA, PDGFRB, or FGFR1
♦
The 2008 WHO classification has placed this group of disorders together as a separate class of entities. Within this group are several rare entities including chronic myelomonocytic leukemia (CMML) with eosinophilia, most cases of chronic eosinophilic leukemia, and cases of T-cell lymphoblastic leukemia with eosinophilia. This group of disorders is characterized by molecular evidence of abnormalities of three genes PDGFRA, PDGFRB, and FGFR1. One of the more common abnormalities is the translocation of the FIP1L1/PDGFRA, which can be tested by FISH studies (by testing for deletions of CHIC2). However, abnormalities of PDGFRB and FGFR1 are often cryptic and can only be evaluated by molecular methods. Those disorders with PDGFRA and PDGFRB can be exquisitely responsive to anti-tyrosine kinase medications such as imatinib, making their identification as a unique clinical subset important
Chronic Eosinophilic Leukemia (Fig. 17.4)
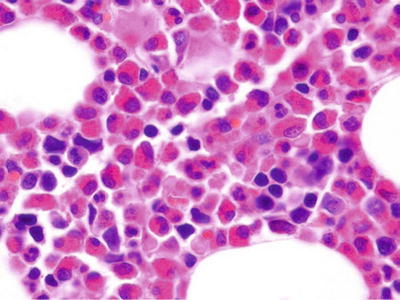
Fig. 17.4.
Chronic eosino philic leukemia. This bone marrow core biopsy shows a massive increase in eosinophils and precursors.
Definition
♦
Autonomous proliferations of eosinophils with associated end-organ damage. Chronic eosinophilic leukemia (CEL) has proven clonality or cytogenetic abnormality; a subset of these cases are classified within the group of disorders with PDGFRA, PDGFRB, or FGFR1 abnormalities
Clinical
♦
CEL is a diagnosis of exclusion; other causes of eosinophilia (allergy, parasites, drug effects, secondary to neoplasms) must be excluded. The M:F ratio is 9:1. Eosinophilic infiltration may lead to damage in the lungs, heart, skin, and GI tract. Constitutional symptoms such as diarrhea and pruritis are common
Diagnosis
♦
Peripheral blood
Eosinophilia of greater than 1.5 × 109/L is seen. Eosinophils may be dysplastic in appearance (abnormal granulation, hyperlobated), but these findings can be seen in reactive eosinophilia
♦
Aspirate smear
Increased eosinophils and precursors are seen. Blasts are <20% (or a diagnosis of AML is made). Charcot–Leyden crystals (condensed aggregates of eosinophilic proteins in a large crystalline shape) may be seen. Dysplastic features of other cell lineages may be seen. Eosinophils may be abnormally chloroacetate esterase positive
♦
Biopsy
Marrow biopsies are hypercellular, predominantly due to increased eosinophils
Additional Studies
♦
Cytogenetics/molecular: 5q33 abnormalities associated with PDGFB; +8; 8p11 associated with FGFR1; t(8;13), t(6;8). Cryptic del(4)(q12) resulting in FIP1L1/PDGRFA gene fusion (note: this abnormality is tested for by deletion of the CHIC2 gene which is lost)
Differential Diagnosis
♦
AML inv(16) – differentiate by cytogenetics and blast count; other MPNs
Prognosis and Treatment
♦
Imatinib, most often used in CML, has activity in some cases of CEL, including some with abnormalities of PDGFRA, PDGFRB, and FGFR1
Chronic Neutrophilic Leukemia
Definition
♦
Very rare disorder which consists of sustained, clonal proliferation of mature neutrophils that lacks BCR/ABL1 and other causes of neutrophilia (e.g., paraneoplastic)
Clinical
♦
There is a possible association with plasma cell myeloma (PCM ). Hepatosplenomegaly is seen in almost all cases. It is an exceedingly rare disorder
Diagnosis
♦
Peripheral blood
Numerous mature neutrophils (>25 × 109/L) are seen. Immature granulocytes number <10%. Dysplasia of neutrophils is not seen. Blasts are not increased and are <1% of the white blood cell count
♦
Aspirate smear
Increased neutrophils are seen. No dysplasia of neutrophils or other lineages is seen. Blasts are not increased and account for <5% of the marrow cellularity
♦
Biopsy
The core biopsy is hypercellular, with sheets and aggregates of neutrophils and precursors. The M:E ratio is typically >20:1. Fibrosis is uncommon
Additional Studies
♦
Cytogenetics/molecular: No specific genetic abnormalities have been identified. Most cases (>90%) have no abnormalities by routine cytogenetics. However, a subset of cases have been identified which have JAK2 mutations or the more recently identified CSF3R mutation. It should be noted that there are no findings of abnormalities associated with other myeloid disorders such as BCR/ABL1, PDGFRA, etc
Differential Diagnosis
♦
CML, CMML, and MDS
MPN, Unclassifiable
Definition
♦
Cases that have clinical or morphologic features of an MPN but lack features of a specific entity or have significant overlap between more than one entity AND lack evidence of BCR/ABL1
Other
♦
Features are variable
♦
May represent very early stages of PV or PMF
♦
Features of MDS are lacking
Mastocytosis (Fig. 17.5)
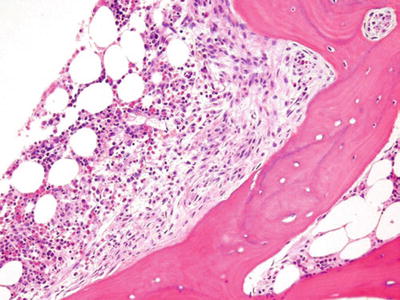
Fig. 17.5.
Mastocytosis. This core biopsy shows a classic paratrabecular aggregate of fibrosis with mast cells. Tryptase, CD 117, Giemsa, or toluidine blue stains would highlight the mast cells.
Definition
♦
Abnormal clonal proliferation and accumulation of mast cells in multiple sites. Disorders included within this diagnosis include cutaneous mastocytosis, indolent systemic mastocytosis, systemic mastocytosis with associated clonal hematologic disorder, aggressive systemic mastocytosis, mast cell leukemia, mast cell sarcoma, and extracutaneous mastocytoma. Cutaneous mastocytosis and extracutaneous mastocytoma will not be discussed further in this section. The reader is referred to appropriate sections in this text
♦
Mast cell leukemia is a very rare manifestation of mastocytosis with >10% mast cells in peripheral blood and diffuse infiltration of the marrow by >20% mast cells
Clinical
♦
Constitutional symptoms may be prominent as well as mast cell cytokine-mediated symptomatology. Elevated serum tryptase levels are almost always seen but are not entirely specific for mastocytosis. Other common sites of involvement in systemic mastocytosis include the spleen, skin, and GI tract. Hepatosplenomegaly is a common clinical finding. Skeletal abnormalities with osteolytic lesions or pathologic fractures are not uncommon
♦
Mastocytosis can be seen in association with other hematopoietic malignancies including MPN, MDS, AML, lymphomas, or other hematologic disorders
Diagnosis
♦
Peripheral blood
Circulating mast cells are rarely seen except in mast cell leukemia. When seen, they may appear similar to monocytes (when degranulated) or may have a more typical mast cell appearance
♦
Aspirate smear
Mast cells are seen in increased numbers. In comparison with normal mast cells, neoplastic mast cells may be hypogranular or spindle shaped. Increased numbers of eosinophils, lymphocytes, or plasma cells may be seen adjacent to or associated with mast cells. Because of the association with other hematopoietic disorders, care must be taken when evaluating other marrow elements
♦
Biopsy
Mast cells are round with moderate amounts of cytoplasm or occasionally appear as unremarkable spindle cells. On H&E stains, mast cells are inconspicuous; cytoplasmic granules may be inapparent. Giemsa or toluidine blue stains may highlight metachromatic granules of mast cells
Mast cell infiltrates are associated with polymorphous infiltrates of inflammatory cells including lymphocytes, plasma cells, and often prominent eosinophils
Fibrosis is common and the classic appearance is of fibrotic paratrabecular aggregates with mast cells and mixed inflammatory cells
Additional Studies
♦
Flow: Often noncontributory due to associated fibrosis, mast cells are positive for CD117, CD45, CD11c, CD33, CD43, CD2, and CD25
♦
IHC: Mast cells are positive for tryptase, CD117, CD43, CD2, and CD25
♦
Note: CD2 and CD25 expression is seen in neoplastic mast cells and not normal mast cells
♦
Cytogenetics/molecular: Activating mutations of the KIT gene can be identified in many cases
Differential Diagnosis
♦
Although there may be overlap, disorders with PDGFRA, PDGRFB, and FGFR1 should be considered separately from cases of mastocytosis
Acute Myeloid Leukemia
AML: General (Fig. 17.6)
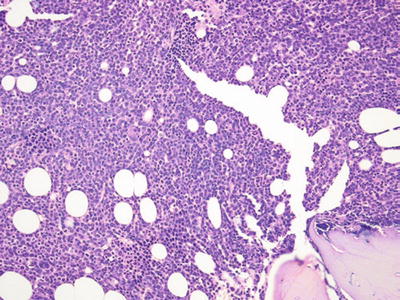
Fig. 17.6.
Acute myeloid leukemia. This core biopsy shows extensive infiltration by myeloid blasts.
Classification
♦
Malignancy of myeloid progenitor/early precursor cells. Several subtypes exist which are based on cytogenetic findings, cellular lineage, and phenotypic variation. The current WHO classification divides acute myeloid leukemia (AML ) into four general groups: AML with recurrent cytogenetic abnormalities, AML with myelodysplasia-related changes, AML with therapy-related myeloid neoplasms (including AML and MDS), and AML not otherwise categorized
♦
AML, not otherwise categorized includes several clinical and/or morphologic entities, which make up the majority of cases of AML
♦
WHO terminology is used in this section, with the outdated French–American–British (FAB) classification system terminology included in parentheses for historical purposes
Clinical Findings
♦
AML is most often a disease of adults, 60 years and older. Contributing etiologies include certain viruses, ionizing radiation, cytotoxic chemotherapy, benzene, and cigarette smoking
Diagnosis
♦
Auer rods are a fusion of primary granules, seen in the cytoplasm of blasts of granulocytic lineage; they are seen exclusively in a subset of cases of AML or high-grade MDS
♦
Cytochemical stains provide information on lineage of some AML types (see Table 17.1); for myeloperoxidase (MPO) or nonspecific esterase (NSE) to be considered positive, at least 3% of the neoplastic cells (e.g., blasts) must stain positively. While of some benefit, many of the results of cytochemical stains are less important for classification than genetic or immunophenotypic findings
Table 17.1.
Cytochemical Stains Useful in the Diagnosis of Hematopoietic Disorders
Stain | Disorder | Staining pattern | Comment |
|---|---|---|---|
Myeloperoxidase (MPO) | AML, myeloblastic AML, myelomonocytic APML | >3% blasts stain positive | |
Sudan black B (SBB) | AML, myeloblastic AML, myelomonocytic APML | Results similar to those of MPO | |
Nonspecific esterase (NSE) | AML, monocytic AML, myelomonocytic AMKL | >3% of monoblasts or promonocytes stains positive | |
Chloroacetate esterase (CAE) | AML, myeloblastic APML | Less sensitive than MPO | Highlights Auer rods |
PAS | ALL Erythroleukemia | Block positivity. Perinuclear globules | |
TRAPa | Hairy cell leukemia | Abnormal lymphocytes are positive | Some SMZL may also be positive |
AML with Recurrent Cytogenetic Abnormalities
♦
In cases with recurrent cytogenetic abnormalities, even if blast percentages do not reach 20%, they should still be diagnosed at AML rather than MDS
AML with t(8;21)(q22;q22)
♦
Translocation results in fusion of RUNX1/RUNX1T1 (AML1/ETO) genes. Represents 5–10% of cases of AML. Patients are typically younger than in other types of AML
♦
Morphology is most often large blasts, most often with numerous cytoplasmic granules and frequent Auer rods. Maturing neutrophilic elements with or without dysplasia may be seen. Strong cytoplasmic positivity for myeloperoxidase is usually seen
♦
Flow: typically positive for CD34, CD13, and CD33 (often weak) and often coexpresses CD19, with variable CD56
♦
Prognosis is good, compared to other types of AML
AML with inv(16)(p13.1q22) or t(16;16) (p13.1;q22)
♦
Most commonly results in fusion of CBFB/MYH11 genes. Accounts for 10–12% of cases of AML and often has features of myelomonocytic leukemia. Most cases have atypical eosinophil precursors with heavy granulation and abnormally large granules. Eosinophilia may or may not be present in the peripheral blood. Cytochemistry for myeloperoxidase is typically positive in blasts (at least 3%) and a component of monoblasts/promonocytes that are positive for NSE (see Table 17.1)
♦
Flow: Component of myeloid blasts positive for CD13, CD117, CD15, CD33, and myeloperoxidase. The monocytic elements are variable positive for CD11b, CD4, CD14, and CD64. CD2 is often aberrantly expressed on myeloid component
♦
Prognosis is good, compared to other types of AML
AML with t(9;11)(p22;q23)
♦
Translocation of MLLT2/MLL genes. Occurs more frequently in AML in infants (9–12%) and fewer adults AMLs (2%). It is also seen in association with therapy-related AML (see later section). Monocytic morphology is typical, or less commonly, myelomonocytic features are seen. Increased incidence of extramedullary involvement (e.g., myeloid sarcoma) is seen
♦
NSE positivity by cytochemistry is seen in most cases
♦
Flow: There are no specific flow findings for AML with 11q23 abnormalities, but monocytic lineage markers (CD11b, CD4, CD14, CD64) are often positive
♦
Prognosis of t(9;11) is poor
AML with Alternate MLL Translocations
♦
Translocation of MLL genes with several other partner genes including MMLT3, MLLT1, MLLT10, EL L, and others [t(11;19); t(4;11), etc.]
♦
Occurs more frequently in therapy-related AML (see later section)
♦
Monocytic morphology is typical
♦
Prognosis of MLL gene translocations in AML is typically poor
Acute Promyelocytic Leukemia (Fig. 17.7)
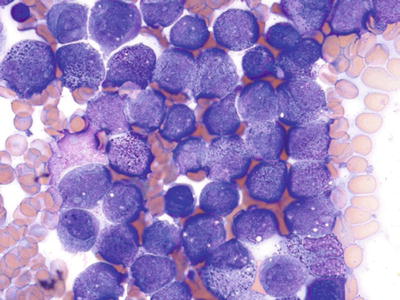
Fig. 17.7.
Acute promyelocyte leukemia. This aspirate smear shows a large cluster of abnormal, granular promyelocytes.
Definition
♦
An acute myeloid leukemia with a predominance of neoplastic promyelocytes. There are two morphologic variants: hypergranular or “typical” type and the microgranular or hypogranular type
Clinical
♦
Clinical ly can be associated with disseminated intravascular coagulation, making this diagnosis a potential medical emergency. This subtype represents 5–8% of all cases of AML
♦
(FAB: AML-M3)
Diagnosis
♦
Peripheral blood
Microgranular variant typically has few cells with identifiable granules (granules are present, but smaller than that can be resolved by the eye – they can be seen by electron microscopy). Typical acute promyelocytic leukemia (APML) has hypergranular promyelocytes, often numerous in peripheral blood smear. Neoplastic promyelocytes are intermediate in size with irregular nuclei. Nuclei often have a “figure eight” or “ dumbbell” shape. Auer rods may be seen; cells with numerous Auer rods are termed “faggot cells.”
♦
Aspirate smear
Abnormal promyelocytes account for at least 20% of cells. Promyelocytes are intermediate-sized cells with moderate amounts of cytoplasm and numerous granules; Auer rods may be present. Nuclear irregularities are common. Myeloperoxidase cytochemistry strongly stains abnormal promyelocytes
♦
Biopsy
The core biopsies are typically hypercellular. Normal marrow elements are replaced by promyelocytes. These neoplastic promyelocytes often have irregular, folded nuclear contours and moderate amounts of pink cytoplasm
Additional Studies
♦
Flow: Immunophenotype is characteristic but not entirely specific for APML. Phenotype is positive for myeloid markers CD13 and CD33 and often weak for CD117. They are almost always negative for CD34 and HLA-DR (in contrast to most other AMLs). About 20% of cases express CD56
♦
Cytogenetics/molecular: Classic translocation t(15;17) (q22;q12) – PML/RARA; other translocations can occur but are rare – t(11;17); t(5;17)
Differential Diagnosis
♦
Other types of AML
Prognosis and Treatment
♦
Favorable prognosis AML
♦
Treatment includes the use of all-trans-retinoic acid (ATRA), which causes differentiation of neoplastic cells. t(11;17) variant appears to be resistant to ATRA
AML with t(6;9)(p23;q34)
♦
Translocation of DEK/NUP214 genes. Represents 0.7–1.8% of AML and occurs in both children and adults. Monocytic morphology is typical, and basophilia and multilineage dysplasia are also frequently seen
♦
AML with t(6;9) is associated with a poor prognosis
AML (and MDS), Therapy Related
Alkylating Agent-Related AML
♦
Latency is usually 5–6 years after treatment. This type of AML often arises from a previous MDS or is associated with significant dysplastic features. Morphology may be myeloid, monocytic, myelomonocytic, or other AML types. Marrow fibrosis is not uncommon
♦
Flow: Abnormal blast population will be present and often expresses CD34, other myeloid markers (CD13, CD33), and possibly monocytic markers (CD64, CD14), with variable expression of CD56 and CD7
♦
Genetics: These cases often have complex cytogenetics and may include MDS-like changes including deletions/monosomies of chromosomes 5 and 7
♦
Prognosis: Overall, the prognosis is poor. However, therapy-related AML with t(15;17) appears to have a similarly good prognosis to de novo cases
Topoisomerase Type II Inhibitor-Related AML
♦
Latency is approximately 3 years after treatment. Drugs associated with this type of leukemia include epipodophyllotoxins, etoposide, and teniposide. Usually there is no preceding MDS phase. Blasts are monocytic or have some evidence of a monocytic differentiation
♦
Cytogenetics: In this AML, there are typically abnormalities that involve the MLL gene at 11q23 including translocations t(11;19), t(9;11), and t(4;11)
AML with Gene Mutations
♦
Specific gene mutations have been identified that are both associated with chromosomal translocations and independent of those changes. Mutations in FLT-3, NPM1, CEBPA, C-KIT, and WT1 are all associated with AML. Each change may be associated with specific prognostic implications. Most importantly, individual prognosis and therapy of AMLs will likely be based on these and other molecular subclassifications in the future:
NPM1: Normal cytogenetics. Favorable prognosis
CEBPA: Normal cytogenetics. Favorable prognosis
FLT-3: Normal cytogenetics. Most common molecular abnormality identified; internal tandem duplications (FLT-3-ITD) are associated with an adverse prognosis
AML with Myelodysplasia-Related Changes
♦
AML with a prior history of MDS or morphologic features of dysplasia. There is also an absence of previous cytotoxic therapy or findings associated with specific genetic changes of another AML type
♦
Morphologic features are those of MDS with >20% blasts in the peripheral blood or bone marrow
♦
Prognosis is poor
AML, Not Otherwise Categorized
♦
Listed below, these leukemias are classified based on morphologic, immunophenotypic, or clinical findings and lack features of other types of leukemias
♦
This large group accounts for the majority of cases of de novo AML
AML: Myeloblastic
Definition
♦
As a group, these are acute leukemias with varying degrees of granulocytic lineage differentiation
♦
Acute myeloblastic leukemia, with minimal differentiation (FAB: AML-M0)
♦
Acute myeloblastic leukemia, without maturation (FAB: AML-M1)
♦
Acute myeloblastic leukemia, with maturation (FAB: AML-M2)
Clinical
♦
Combined, these groups account for 45–60% of AMLs. Typical symptoms of AML are usually present
Diagnosis
♦
Peripheral blood
Varying numbers of blasts are seen. Blasts may be undifferentiated or have unequivocal features of myeloblasts including cytoplasmic granules occasionally with Auer rods
♦
Aspirate smear
Blasts comprise at least 20% but are often more numerous. Cytochemistry in minimal differentiation has no positivity for myeloperoxidase. In cases without maturation and with maturation, myeloperoxidase is positive in at least 3% (but usually more) blasts (see Table 17.1)
Minimally differentiated: Blasts are intermediate to large in size with open, delicate chromatin and small amounts of agranular cytoplasm
Without maturation: Blasts often account for >90% of the nonerythroid cells present; no myeloid maturation is identified
With maturation: Blasts account for at least 20% of the cells present, at least 10% of the cells present display neutrophilic maturation, with <20% monocytic lineage cells; dysplastic features may be present in the neutrophilic series
♦
Biopsy
Typically hypercellular with a prominent component of blasts present
Additional Studies
♦
Flow
Minimally differentiated cases are positive for at least one panmyeloid antigen such as CD13, CD33, or CD117. They are negative for B or T antigens. TdT is positive in about one-third of cases. Flow methods for myeloperoxidase are more sensitive than cytochemistry and are positive even in cases with negative cytochemistry
In AML without and with maturation, the immunophenotype is typically positive for CD34, CD13, and CD33 with variable CD117
♦
IHC: These cases are often positive for CD34, but this marker lacks lineage specificity
Prognosis
♦
AML with minimal differentiation is associated with a poor prognosis
♦
The prognosis is variable in AML with and without maturation. Their prognosis is dependent on underlying molecular and cytogenetic abnormalities present
AML: Monocytic/Monoblastic (Fig. 17.8)
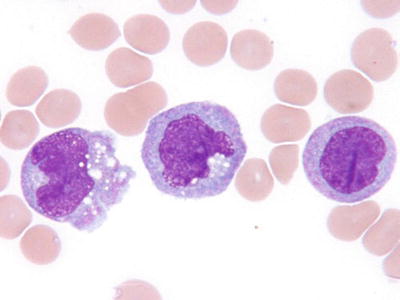
Fig. 17.8.
AML, with monocytic/monoblastic differentiation. This peripheral blood smear illustrates monocytic differentiation in circulating blasts of AML.
Definition
♦
An acute leukemia where >80% of the leukemic cells are of monocytic lineage; this includes monoblasts, promonocytes, and mature monocytes. Neutrophils and precursors should be <20% (see AML, myelomonocytic)
Clinical
♦
Monocytic or monoblastic AMLs represent 3–6% of all cases of AML. Extramedullary disease, including gum infiltrates, and cutaneous and CSF involvement are common
♦
FAB: AML-M5a – monoblastic; FAB: AML-M5b – monocytic
Diagnosis
♦
Peripheral blood
Blasts are often present and varying from poorly differentiated to those with a clear monocytic appearance. Blasts of monocytic lineage often have nuclear folds or irregularities and increased amount of blue or blue-gray cytoplasm and lack cytoplasmic granules
♦
Aspirate smear
Blasts or blast equivalents (promonocytes) in aspirate (or PB) are greater than 20%. Monocytic differentiation may be less apparent in aspirate than in peripheral blood. Promonocytes (immature monocytic forms) are counted as blasts but may be difficult to distinguish from other cell types unless numerous. By cytochemistry, nonspecific esterase is typically positive in monoblasts and promonocytes (see Table 17.1)
♦
Biopsy
Biopsy is usually hypercellular with numerous blasts. Promonocytes have somewhat folded irregular appearance of nuclei
Additional Studies
♦
Flow: Blast population variably expresses monocytic/myeloid antigens including CD11b, CD14, CD4, and CD64. CD117 expression is not uncommon. CD34 is less commonly positive in the neoplastic population
♦
IHC: Monocyte/macrophage markers (e.g., CD68, CD163, lysozyme) may be negative in less differentiated forms of monocytic/monoblastic leukemia
♦
Cytogenetics/molecular: Cytogenetics abnormalities vary
Differential Diagnosis
♦
AML with monocytic differentiation and 11q23 (MLL) abnormalities should be classified with “AML with recurrent cytogenetic abnormalities” group
AML: Myelomonocytic (Fig. 17.9)
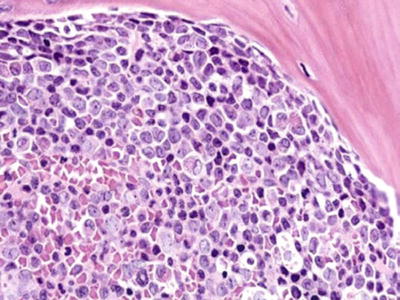
Fig. 17.9.
AML, myelomonocytic. This core biopsy is infiltrated by blasts, some with irregular folded nuclei, suggesting monocytic differentiation.
Definition
♦
An acute leukemia with proliferation of both monocytic and neutrophil precursors
Clinical
♦
This subtype accounts for 15–25% of cases of AML. Myelomonocytic leukemia may have extramedullary manifestations similar to monocytic/monoblastic leukemias
♦
(FAB: AML-M4)
Diagnosis
♦
Peripheral blood
Often large numbers of circulating monocytes are seen. Circulating (undifferentiated) blasts are commonly seen
♦
Aspirate smear
At least 20% blasts (or blasts + promonocytes) are seen. In addition, there must be at least 20% monocytes and monocytic precursors and at least 20% neutrophils and precursors
By cytochemistry, distinct populations of myeloperoxidase-positive and nonspecific esterase-positive blasts are present, representing the myeloblastic and monoblastic components, respectively (see Table 17.1)
♦
Biopsy
The core biopsy is typically hypercellular. Blast population is often large and easily identifiable
Additional Studies
♦
Flow: Immunophenotype by flow will typically show a component of myeloblasts (CD13, CD33, CD34) and monoblasts (CD11b, CD4, CD64, CD117)
♦
Cytogenetics/molecular: Abnormalities are often present but nonspecific
Differential Diagnosis
♦
AML with inv(16)
Acute Megakaryoblastic Leukemia (Fig. 17.10)
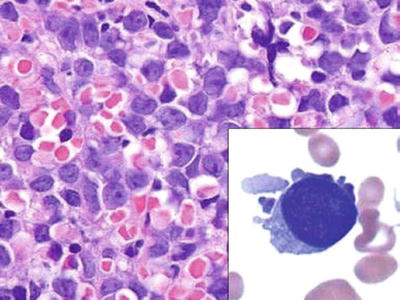
Fig. 17.10.
Acute megakaryoblastic leukemia. The core biopsy is infiltrated by large, undifferentiated blasts. Inset: a single megakaryoblast, with cytoplasmic blebs.
Definition
♦
Leukemic proliferation of blasts, with > 50% being of megakaryocytic/megakaryoblastic lineage
Clinical
♦
This represents 3–5% of all cases of AML. Increased incidence seen in patients with Down syndrome (trisomy 21)
♦
(FAB: AML-M7)
Diagnosis
♦
Peripheral blood
Circulating blasts often present. Abnormal or bizarre platelets or micromegakaryocytes may be seen
♦
Aspirate smear
Blasts are of medium–large size and the classic appearance of megakaryoblasts includes deep blue “blebs” of cytoplasm (ineffective formation of platelets)
♦
Biopsy
The core biopsies are hypercellular. The cellularity is often composed of a uniform population of poorly differentiated, relatively large blasts. Severe reticulin fibrosis is common. Rarely, the presence of more mature, identifiable megakaryocytic forms is present
Additional Studies
♦
Flow: Megakaryoblasts express CD41 and/or CD61. Other myeloid markers vary
♦
IHC: Useful markers for the identification of megakaryoblasts include factor VIII, CD61, and CD42b
♦
Cytogenetics/molecular: Trisomy 21 and inv(3) can be seen, but no changes are specific for acute megakaryoblastic leukemia. iso(12p) is seen exclusively in patients with mediastinal germ cell tumors with a component transforming to AML
Differential Diagnosis
♦
Transient myeloproliferative disorder of Down syndrome (TMPD): Immature cells and blasts in peripheral blood of Down syndrome patients; most cases remit spontaneously, while others progress to leukemia; Most cases have GATA-1 mutations
Acute Erythroid Leukemia (Fig. 17.11)
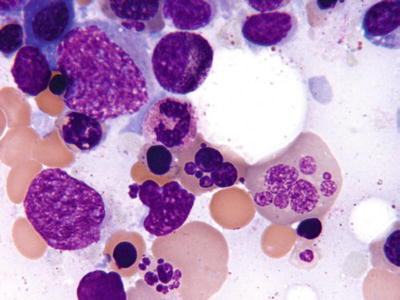
Fig. 17.11.
Erythroleukemia . This aspirate smear shows the severe erythroid dysplasia often seen in erythroleukemia.
Definition
♦
Very rare acute leukemia that consists of two main subtypes:
Erythroleukemia is characterized by the presence of >50% erythroid precursors of all nucleated cells AND >20% myeloblasts of all nonerythroid cells present
Pure erythroid leukemia is characterized by a marrow consisting of >80% immature erythroid cells without significant numbers of myeloblasts
Clinical
♦
Profound anemia is common. There is considerable diagnostic overlap with AML with myelodysplasia-related changes. It represents <5% of all types of AML. Pure erythroid leukemia is extremely rare (<1%)
♦
(FAB: AML-M6)
Diagnosis
♦
Peripheral blood
Severe RBC anisopoikilocytosis is not uncommon. A component of circulating blasts may be present
♦
Aspirate smear
Severe erythroid dysplasia is almost always present. Erythroid dysplasia includes megaloblastoid chromatin, multinucleation, nuclear fragmentation, giant erythroblasts, and cytoplasmic vacuolation. Immature erythroid cells are increased. More common type (erythroleukemia) includes component of typical myeloblasts (often with myeloperoxidase positivity)
Cytochemistry in acute erythroid leukemia may highlight vacuoles in abnormal erythroid precursors with strong, abnormal PAS positivity. An iron stain may highlight ringed sideroblasts
♦
Biopsy
The marrow cores are hypercellular, with increased erythroid elements. The amount and distribution of blasts are variable depending on the type of acute erythroid leukemia. Core biopsy morphology of immature erythroid elements can mimic other disorders and should be confirmed by smear morphology or immunophenotypic confirmation
Additional Studies
♦
Flow: Prominent populations of erythroid cells are present (markers such as bright CD71 may confirm erythroid lineage). Myeloblasts, with typical myeloid markers (CD13, CD33, CD34, CD117), are variably present
♦
IHC: CD71, E-cadherin, anti-hemoglobin, or anti-glycophorin antibodies are often useful in identifying immature erythroid cells
♦
Cytogenetics/molecular: Complex karyotypes are common. Abnormalities of chromosomes 5 and 7 are fairly frequent
Prognosis
♦
Often aggressive disease with a poor prognosis
Acute Panmyelosis with Myelofibrosis (Fig. 17.12)
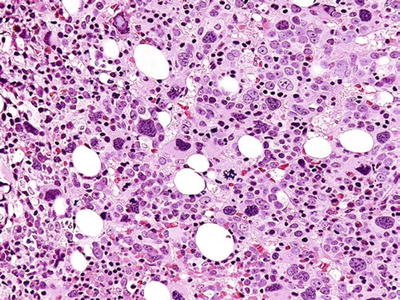
Fig. 17.12.
Acute panmyelosis with myelofibrosis. This core biopsy is hypercellular, with abnormal-appearing megakaryocytes and a population of blasts. Extensive fibrosis is present, and the lineage of the blasts is usually characterized by immunohistochemistry.
Definition
♦
AML subtype with proliferation of multilineage blasts and significant bone marrow fibrosis
Clinical
♦
Acute panmyelosis with myelofibrosis (APMF ) is very rare and accounts for <1% of all cases of AML. There is no or minimal splenomegaly, in contrast to PMF, an important differential diagnostic consideration. This entity is distinct from acute megakaryoblastic leukemia (see above) although historically cases may have been misclassified. This disorder is often associated with a rapid course and dismal prognosis. Previous names include acute myelofibrosis and acute myelos clerosis
Diagnosis
♦
Peripheral blood
Often pancytopenia is seen. Occasional circulating blasts may be seen. Typically there is a lack of anisopoikilocytosis of red blood cells
♦
Aspirate smear
Aspirates are typically hypocellular and hemodilute, as well as inaspirable due to reticulin fibrosis
♦
Biopsy
The cores are hypercellular, with abnormal proliferations of myeloid, erythroid, and megakaryocytic lineages. Dysplasia of all lineages is typically present but may be most prominent in megakaryocytes. Immature hematopoietic cells are present and represent immature myeloid, erythroid, and megakaryoblastic elements (as identified by IHC). Severe fibrosis is present in all cases
Additional Studies
♦
Flow: Not usually available due to lack of aspirate; if present may highlight increased myeloblasts
♦
IHC: CD34 is positive in subpopulation of blasts, mostly the myeloblasts. Some immature cells of erythroid lineage are identified by staining for CD71, E-cadherin, or other erythroid markers. A component of immature cells of the megakaryocytic/blastic lineage can be identified by staining for factor VIII, CD31, CD61, and CD42b
Differential Diagnosis
♦
AMKL, AML with fibrosis, or MDS with fibrosis
Prognosis
♦
Very poor prognosis, despite treatment
Other Forms of AML
♦
Acute basophilic leukemia: very rare and blasts >20% with evidence of basophilic differentiation
Acute Leukemias of Ambiguous Lineage
Definition
♦
Acute leukemias which have morphologic, cytochemical, or immunophenotypic features that either (1) lack clear features of either myeloid or lymphoid origin or (2) have characteristics of both myeloid and lymphoid lineages or both B-cell and T-cell lineages
♦
Myeloid lineage is defined as either evidence of myeloperoxidase expression (by flow, IHC, or cytochemistry) or monocytic differentiation (cytochemical expression of NSE or phenotypic expression of CD11c, CD14, and CD64 or expression of lysozyme)
♦
T-cell lineage is defined as expression of cytoplasmic CD3 (by flow or other methods) or surface CD3 expression (which is rare in leukemias of ambiguous lineage)
♦
B-cell lineage is characterized as expression of strong CD19 with strong expression of other B-cell antigens, such as CD79a and cytoplasmic CD22, or CD10 expression or weak expression of CD19 with strong expression of at least two additional antigens (CD79a, cytoplasmic CD22, and CD10)
Acute Undifferentiated Leukemia
♦
Lacks lineage specific markers for B-ALL, T-cell lymphoblastic leukemia/lymphoma (T-ALL), and AML
Mixed Phenotype Acute Leukemia with t(9;22) (q34;q11.2)
♦
This diagnosis is observed in patients with an acute leukemia and no known previous history of CML. These will typically have a dimorphic blast population with typical-appearing myeloblasts and lymphoblasts
Mixed Phenotype Acute Leukemia with t(v;11q23) MLL Rearranged
♦
Has other criteria of mixed phenotype leukemia with translocations of the MLL gene
♦
Dimorphic population of blasts is seen
Mixed Phenotype Acute Leukemia, B/Myeloid, NOS
♦
Associated with a poor prognosis
Mixed Phenotype Acute Leukemia, T/Myeloid, NOS
♦
Associated with a poor prognosis
Blastic Plasmacytoid Dendritic Cell Neoplasm
Definition
♦
A clinically aggressive neoplasm of cells derived from a precursor of plasmacytoid dendritic cells, which is a myeloid-related lineage
Clinical
♦
Patients will often have both leukemic disease and cutaneous tumors. Most patients are elderly, and this neoplasm occurs in males more frequently. There is some association with CMML. Some cases will evolve from CMML, and BPDCN is a “blast transformation” event
Diagnosis
♦
Peripheral blood
Frequently peripheral blood appearance is of blasts, with open chromatin, scant cytoplasm, prominent nucleoli, and no or rare cytoplasmic granules
♦
Aspirate smear
Aspirates may show numerous blasts or the infiltrates of neoplastic cells may be subtle
♦
Biopsy
Involvement in the bone marrow can be subtle or massive
Additional Studies
♦
Flow: Immunophenotype of these cells shows coexpression of CD4 and CD56, with expression of CD123. Other markers are variable, but often cases will express CD33 and CD7, with fewer cases expressing CD2 and CD38. There is typically no expression of CD3, CD5, CD13, CD16, CD19, or CD20. CD34 and CD117 are negative and TdT is positive in a subset of cases
♦
IHC: Staining for CD4 and CD56 is of benefit, but expression of CD123 is very helpful as both a specific and a sensitive stain for this neoplasm. TCL-1 is expressed in all cases
Differential Diagnosis
♦
Chronic myelomonocytic leukemia
Prognosis
♦
Very poor prognosis
Acute Lymphoblastic Leukemia
B-Cell Lymphoblastic Leukemia/Lymphoma (Fig. 17.13)
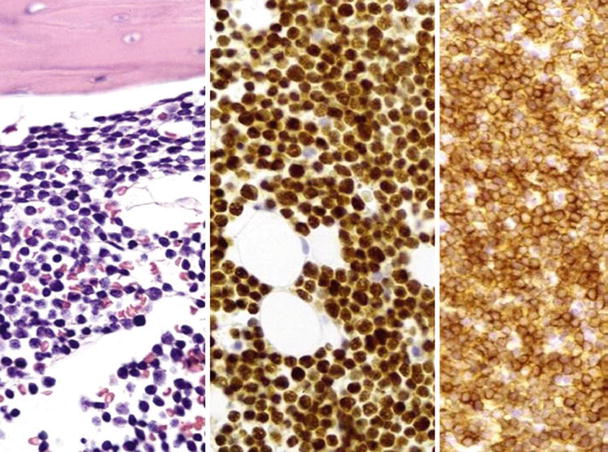
Fig. 17.13.
Precursor B-cell acute lymphoblastic leukemia. This composite photo shows (left to right) core biopsy, TdT staining, and CD79a staining in precursor B-ALL.
Definition
♦
A neoplasm of lymphoid precursor cells (lymphoblasts) of B-cell origin which accounts for approximately ~90% of cases of ALL. There are two main subtypes of B-cell lymphoblastic leukemia/lymphoma (B-ALL ): (1) those with recurrent cytogenetic abnormalities and (2) those without recurrent genetic abnormalities (referred to specifically as B-cell lymphoblastic leukemia, NOS)
Clinical
♦




Common pediatric malignancy and is relatively rare in adults. Initial presentation may present as infection, due to underlying hematologic abnormalities. Extramedullary involvement (CSF, skin, gonads) is common
Stay updated, free articles. Join our Telegram channel
Full access? Get Clinical Tree
