vaso-occlusive crises. Infarction results from vaso-occlusive events (Fig. 12.4). The rigid sickle cells become sequestered by the reticuloendothelial system (spleen), which leads to shortened red cell survival. Massive blood pooling in the spleen can occur.
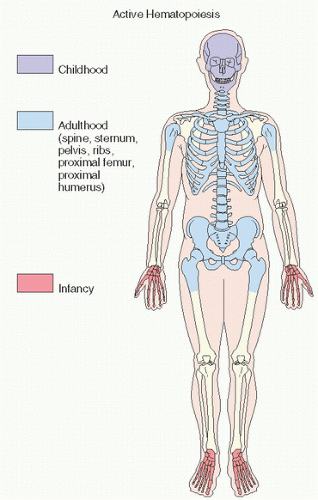 FIGURE 12.1. Sites of active hematopoiesis at different stages of life. |
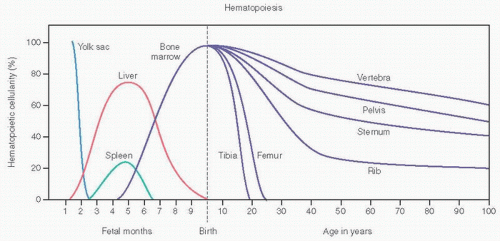 FIGURE 12.2. Hematopoiesis and age. (Adapted from Lukens JN. Blood formation in the embryo, fetus, and newborn. In: Lee CR, et al., eds. Wintrobe’s Clinical Hematology. 9th ed. Philadelphia, PA: Lea & Febiger; 1993:79-100.) |
TABLE 12.1 Normal Hemoglobins (Hb) in Adults | ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
|
growth disturbances can be seen. In one study of 57 sicklers from Saudi Arabia, Bennett and Namnyak (7) detailed the clinical problems (Table 12.3). The hand and foot syndrome, an early manifestation in about one-third of cases, is thought to be a manifestation of bone and marrow infarction.
TABLE 12.2 Percentages of Hemoglobin (Hb) in Normal Persons and Those with Sickle Cell Disease and Sickle Cell Trait | ||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
|
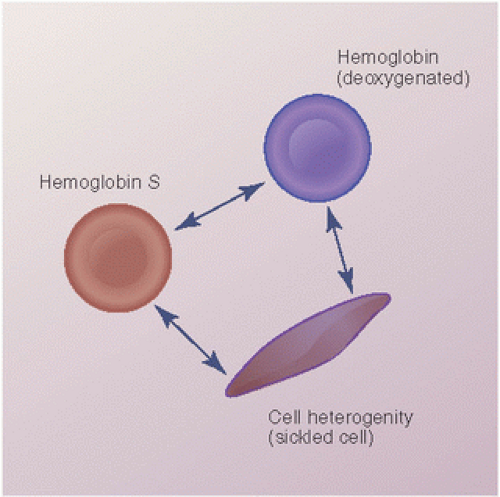 FIGURE 12.3. The progression of hemoglobin S to a deoxygenated state, and its further polymerized state seen as a sickled cell. |
 FIGURE 12.4. Peripheral smears from a normal person (A), a person with iron deficiency (B), a patient with thalassemia (C), and a sickler (D). Normal red blood cells have a slightly ovoid shape with central pallor. In iron deficiency and thalassemia, cells are small (microcytic) and pale and have less hemoglobin (hypochromic). In more severe thalassemia (C), hemolytic changes become apparent, including the presence of target cells and broken, fragmented cells. In (D), the classic crescent-shaped cells of a sickler are evident. |
TABLE 12.3 Clinical Details in 57 Patients with Sickle Cell Anemia | ||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| ||||||||||||||||||||||||||||
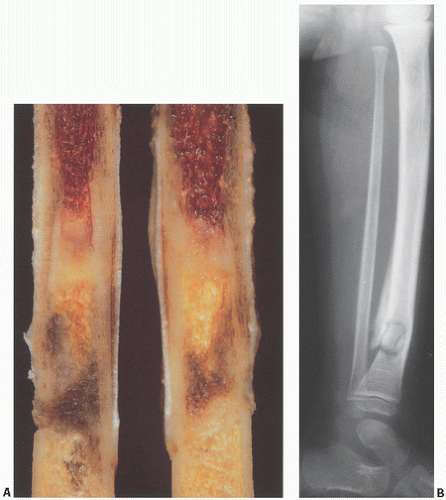 FIGURE 12.5. (A) Sickle cell anemia. Amputated tibia shows viable hyperplastic red marrow proximally and gradations of infarction distally. (B) Osteomyelitis in a case of sickle cell anemia. Areas of sclerosis and lucency in the tibia are surrounded by periosteal reaction. Pathologic fracture is present. The infection was caused by Staphylococcus aureus. |
TABLE 12.4 Characteristics of β-Thalassemias | |||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| |||||||||||||||||||||||||||||||||||
bones is found in thalassemia major. This is most commonly seen in the distal femur, where hyperactive marrow causes widening of the metaphyses and “Erlenmeyer flask” deformity, which can also be found in Gaucher disease, Pyle disease, and osteopetrosis. Similar metaphyseal deformity can be seen in other long bones of patients with these diseases. A “cobwebbing” pattern may be seen in trabecular bone.
 FIGURE 12.6. (A) The pathogenesis of bone and bone marrow changes in β-thalassemia resulting from hemolysis and ineffective erythropoiesis. (Modified from Olivieri, NF. The β-thalassemias. N Engl J Med. 1999;341:99-109.) (B) Thalassemia. Changes in the hand (a) and upper extremity (Continued) |
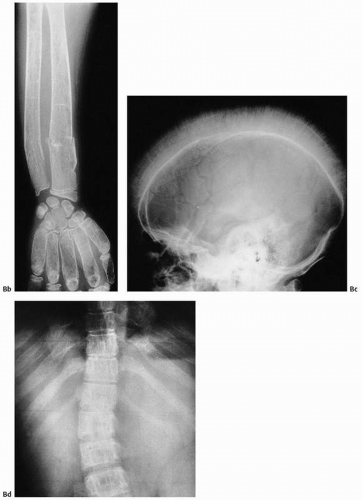 FIGURE 12.6. (Continued) (b) include medullary widening, cortical thinning, marked osteoporosis, and accentuation of the trabeculae of all short tubular bones. The skull may show a hair-on-end appearance (c). In (d), accentuation of trabeculae of the spine and widening of posterior portions of the rib are seen. |
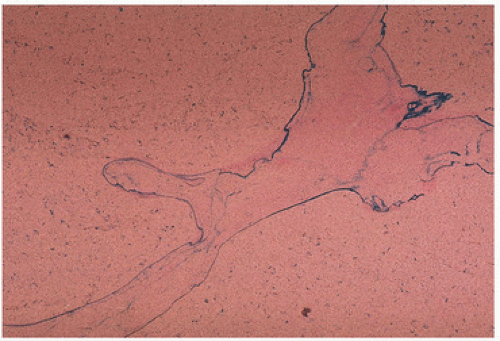 FIGURE 12.7. Thalassemia. Iron (blue) is localized to the mineralizing fronts of bone (Prussian blue stain). |
 FIGURE 12.8. Cystic remodeling of the glenohumeral joint in a patient with hemophilia. (A) Large subchondral erosions are present in the humeral head and glenoid. (B) Computed tomography (CT) shows areas of increased density in the soft tissues, secondary to hemosiderin deposition in the capsule of the joint. (Continued) |
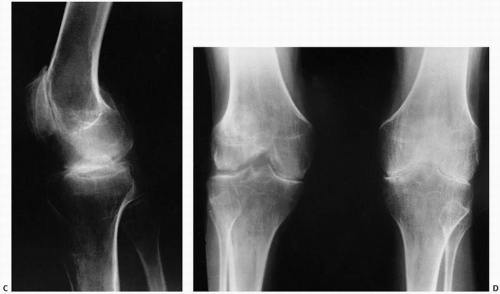 FIGURE 12.8. (Continued) (C, D) Degenerative changes of the knee. |
MRI studies indicate a heterogeneous low-intensity signal on T1-weighted images with a surrounding capsule and septa of low signal intensity and a high-intensity signal on T2-weighted images (27).
 FIGURE 12.9. Roentgenographic (A), gross (B), and histologic (C) appearance of myelofibrosis. Dense bone results from increased bone remodeling associated with paratrabecular fibrosis. The causal relationship is unknown. |
in radiographically evident sclerosis (density) that in its terminal stages causes an inability to extract marrow for diagnosis (“dry tap”). Roentgenographic changes are seen in approximately half the cases and particularly affect sites of adult hematopoiesis (Fig. 12.10).
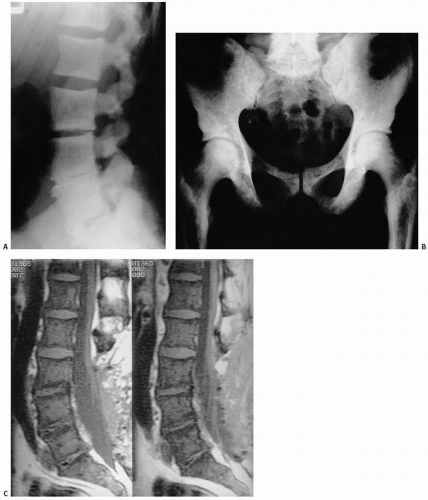 FIGURE 12.10. Radiographs of the spine (A) and pelvis and femora (B) in a patient with myelofibrosis reveal dense (sclerotic) bone. Normal bone contours are essentially maintained in the sagittal MRI. (C) The signal is low on a T1-weighted image because of replacement of the normal marrow fat by fibrosis. |
axial skeleton and large bones are most often affected. Malignancies that result in osteoblastic metastases, such as those of the prostate or the breast, can at times be confused with myelosclerosis.
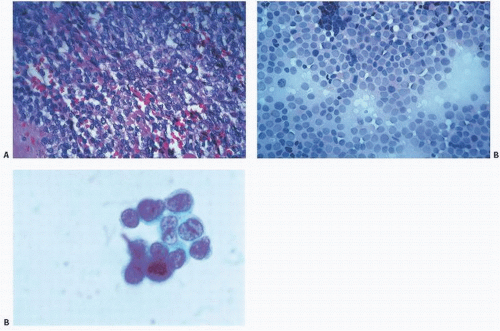 FIGURE 12.11. Small round (blue) cell tumors. “Small” refers to the size of the cells in relation to other cells and to the scant amount of cytoplasm; “blue” to the uptake of hematoxylin stain by the nucleus; “round” to its contour resulting from the nucleus being usually round. (A) Tissue section. (B) Touch imprint. (C) Cytology preparation. Increasingly finer detail is noted with each technique. |
TABLE 12.5 Small Round Cell Tumors of Bone | ||||||
|---|---|---|---|---|---|---|
|
constitutional symptoms such as fever and anemia, weight loss, and leukocytosis are common. Symptoms such as fever, laboratory findings of elevated sedimentation rate, and leukocytosis may lead to a misdiagnosis of osteomyelitis and a potentially catastrophic delay in diagnosis and treatment. A sharp and defined margin best visualized on T1 MRI images in comparison with short tau inversion recovery images has been suggested to be the most significant feature of Ewing sarcoma in differentiating it from osteomyelitis (37).
 FIGURE 12.12. Small round cell tumors. (A) Neuroblastoma (note rosettes). (B) Ewing sarcoma. (C) Well-differentiated lymphocytic (mimics normal lymphocytes) lymphoma. (D) Large cell lymphoma. (E) Embryonal rhabdomyosarcoma (note wisps of red “muscle” cytoplasm). |
Foot to hand, 6:1
Leg to forearm, 69:6
Femur to humerus, 63:32
Pelvic girdle to shoulder girdle, 52:5
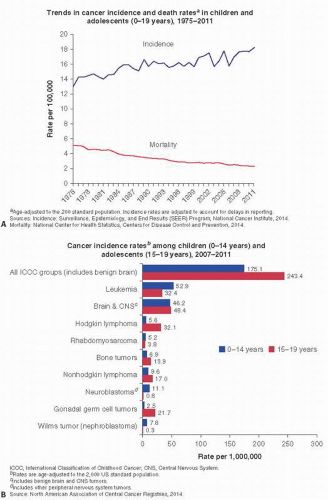 FIGURE 12.13. Cancer Facts (American Cancer Society 2015). |
MRI and PET scanning are better to define the extent of the tumor, with MRI particularly useful in defining marrow extension. MRI may be helpful in monitoring response to chemotherapy. Unusual roentgenographic presentations include cyst-like lesions (41) and those resembling fibrous dysplasia (42).
 FIGURE 12.14. Skeletal distribution of Ewing sarcoma. |
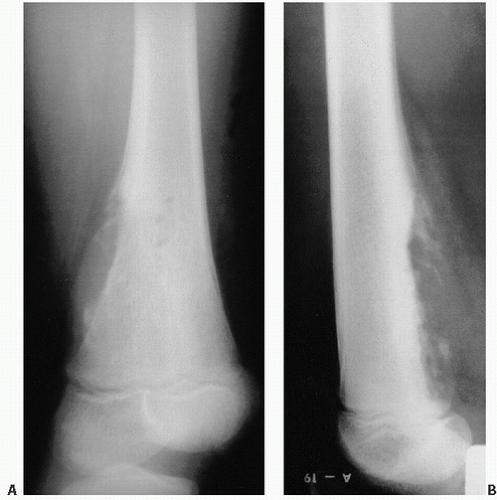 FIGURE 12.15. (A, B) Roentgenographic features of Ewing sarcoma. A lytic lesion is poorly defined, with obvious cortical destruction, soft tissue mass, and periosteal formation of new bone. The tumor is located in the junction of the metaphyses and diaphyses, and apparently spares the growth plate and epiphyses. In most cases, Ewing sarcoma occurs in the diaphyses of long bones and in flat bones. Histology of Ewing sarcoma. (Continued) |
 FIGURE 12.15. (Continued) (C) Sheets of round cells with bland nuclear detail and sparse, indistinct cytoplasm. (D) periodic acid-Schiff stain for glycogen shows abundant pink glycogen in the cytoplasm. (E) Touch preparation. (F) Cytologic detail of fine stippled nuclear chromatin. |
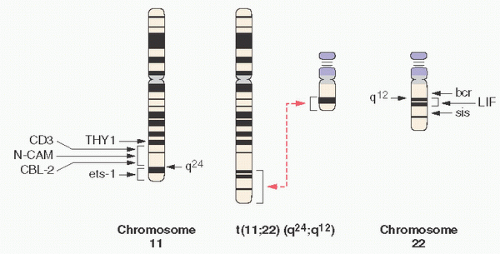 FIGURE 12.16. Translocation t(11;22) in Ewing sarcoma. |
Confirmation at frozen section or biopsy that adequate viable tumor has been sampled
Preparation of cytologic smears or touch imprints
If assessing fluid material (e.g., pleural effusions or tissue from liquefied tumor), the fluid should be centrifuged and the resultant pellet fixed with formalin prior to making a paraffin block. This material can be used for microscopic examination, immunohistochemistry, and polymerase chain reaction (PCR) and fluorescence in situ hybridization (FISH) analyses for cytogenetics.
A minimum of 100 mg of viable tumor snap-frozen for molecular studies
Formalin-fixed tissue for microscopy and immunohistochemistry
Sample fixed in glutaraldehyde for potential ultrastructural studies to identify cross-striations (to rule out rhabdomyosarcoma) or desmosomes (to identify epithelial features)
shown it to be present in other cultured cell lines, including 50 percent of mesenchymal chondrosarcoma, and in some cases of small cell osteosarcoma (52), 90 percent of lymphoblastic lymphomas, 20 to 25 percent of embryonal rhabdomyosarcomas, and 75 percent of poorly differentiated synovial sarcomas. The FLI-1 protein is expressed in 84 percent of Ewing family tumors (33), but may also be seen in lymphoblastic leukemia/lymphoma and other lymphomas. Expression of neural markers such as CD57, S100, and chromogranin may be associated with differentiation toward the PNET end of the Ewing family spectrum. Other markers may be expressed such as keratin (up to 25 percent), vimentin, and desmin, emphasizing the importance of selecting the appropriate antibody panel to cover all entities in the differential diagnosis of these tumors.
TABLE 12.6 Immunohistochemical Profile of Selected Small Cell Tumors | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
0 percent necrosis: No chemotherapy effect = Grade I
<50 percent necrosis: Partial or low chemotherapy effect = Grade IIA
50 percent to 95 percent necrosis: Partial or high chemotherapy effect = Grade IIB
96 percent to 99 percent necrosis: Only scattered viable foci of tumor = Grade III
100 percent necrosis: No residual viable tumor after extensive sampling = Grade IV
Age less than 17
Extremity involvement (distal > proximal)
Appendicular versus axial tumors (56)
Tumors < 8 cm (57)
Nonmetastatic tumors
Type I EWS-FLI1 fusion transcript
>90 percent necrosis following chemotherapy
Patients treated with surgical removal of the involved bone (39)
TABLE 12.7 Poor Prognostic Factors in Ewings Sarcoma | |||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
|
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


