CHAPTER 37 Blood groups on red cells, platelets and neutrophils
Red cell surface antigens
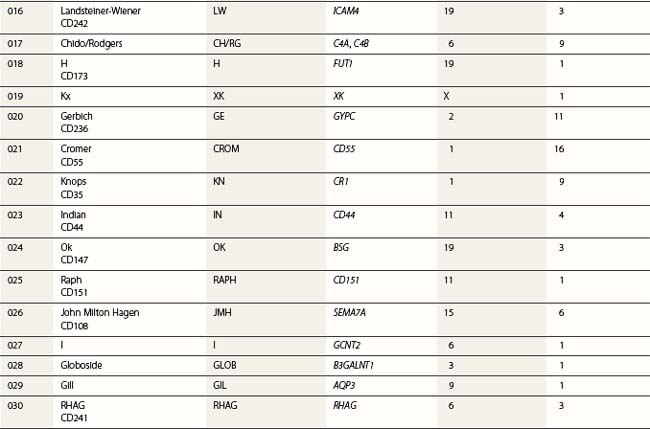
Red cell blood groups have been recognized for over a century, since Landsteiner’s discovery of the ABO system in 1900. Red cell surface antigens are validated and classified by the International Society of Blood Transfusion (ISBT), which currently recognizes 328 antigens, 284 belonging to one of 30 blood-group systems. Each system consists of between one and 52 antigens encoded either by a single gene or by two or three closely linked homologous genes.1,2 The 30 blood group systems are listed in Table 37.1.
Blood-group antigens may be carbohydrate structures on red cell surface glycoproteins or glycolipids, or they may be determined primarily by the amino acid sequence of polypeptides or glycoproteins. At least 23 red cell surface proteins express blood-group polymorphism. The functions of some of these structures are reasonably well understood, such as transport of biologically important molecules in and out of the cell, protection of the cell from autologous complement, and anchoring the membrane to the membrane skeleton. Functions of many, however, can only be speculated on from structural similarity to proteins and glycoprotein of known function.3
Pathology associated with blood-group polymorphism is usually the result of alloimmune destruction of red cells. This might be a hemolytic transfusion reaction following transfusion of red cells to a patient with an alloantibody directed against a determinant present on donor red cells, or hemolytic disease of the fetus and newborn (HDFN), following placental transfer of maternal IgG antibodies into the fetal circulation. Most blood group systems contain a null-phenotype in which the antigens of that system are not expressed. This usually results from an inactivating mutations in the genes encoding the antigen, yet in most cases null-phenotypes are not associated with any pathology, probably due to functional redundancy of cell surface proteins. For reviews on red cell groups see Daniels4 and Reid and Lomas-Francis.5
The ABO and H histo-blood-group systems and other carbohydrate antigens
ABO and H antigens
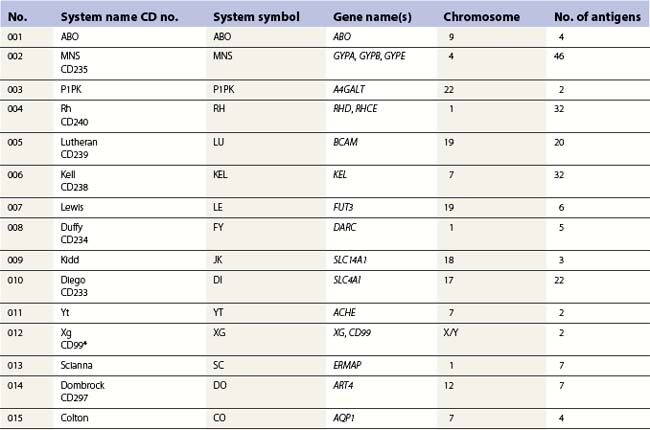
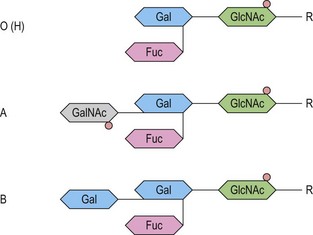
At its most basic level, there are two ABO antigens, A and B, and four phenotypes, A, B, AB and O. O is a null phenotype in which neither A nor B is expressed. The A and B determinants are carbohydrate structures, present on red cell membrane glycoproteins and glycolipids. The major carriers of A and B on red cells are the abundant N-glycosylated glycoproteins, the anion exchanger (band 3) and the glucose transporter (GLUT1). Carbohydrate chains are synthesized by the action of glycosyltransferases, enzymes that catalyse the transfer of specific monosaccharides from a nucleotide donor substrate to an acceptor substrate. The acceptor substrate for A- and B- transferases, products of the A and B alleles, is a terminally fucosylated structure called H antigen (Fig. 37.1). The A gene product is an N-acetylgalactosaminyltransferase that transfers N-acetylgalactosamine from a uridine diphosphate (UDP)-N-acetylgalactosamine donor substrate to the fucosylated galactosyl residue of the H antigen, to produce an A-active structure (Fig. 37.1). B gene product is a galactosyltransferase that transfers galactose from UDP-galactose to the fucosylated galactose of H, to produce B-active structure (Fig. 37.1). The O allele produces no active enzyme, so on group O red cells the H antigen remains unconverted. N-acetylgalactosamine and galactose are the immunodominant sugars of A and B blood groups, respectively.
A- and B-transferases are encoded by a single gene on chromosome 9, cloned by Yamamoto et al6 in 1990. The ABO gene spans about 18–20 kb organized into seven exons of coding sequence. Exons 6 and 7, the two largest, include 77% of the full coding region and encode all of the catalytic domain. The products of the A and B alleles differ by four amino acid substitutions at residues 176, 235, 266 and 268. Leu266Met and Gly268Ala substitutions are most important in determining whether the gene product has predominantly N-acetylgalactosaminyltransferase (A) or galactosyltransferase (B) activity. The sequence of the most common O allele (O1) is identical to the A sequence, apart from a single nucleotide deletion that is responsible for a reading frame shift after codon 86 and the generation of a translation stop signal at codon 117. This allele encodes a truncated protein lacking the catalytic site. There are two other common O alleles: O1v, that, like O1, has the single nucleotide deletion, but differs from O1 by nine nucleotide changes within the coding sequence and O2, that does not have the single nucleotide deletion, but is inactivated by a missense mutation encoding a Gly268Arg substitution (reviewed in 7).
HDFN caused by ABO antibodies
Anti-A and -B are predominantly IgM, but may be IgG. Anti-A,B, which reacts with both A and B antigens, is present in the sera of most group O people and is often partly IgG. ABO HDFN is restricted almost exclusively to group A1 or B fetuses of group O mothers and IgG anti-A,B is generally considered culpable.8
About 15% of pregnancies in women of European origin involve a group O mother with a group A or B fetus. This figure does not vary greatly in most other major ethnic groups, yet ABO HDFN requiring clinical intervention is rare, though minor symptoms involving a small degree of red cell destruction may be relatively common. Hydrops due to ABO HDFN is exceedingly rare, but very occasionally exchange transfusion for the prevention of kernicterus is indicated.8 The main reasons for the low prevalence of clinically significant ABO HDFN is that A and B antigens are present in many tissues. Any antibody crossing the placenta is likely to become bound to placental tissue, reducing the quantity available for destruction of red cells. In addition, immune anti-A,B are mainly IgG2, which does not cause HDFN because there are no Fc receptors for IgG2 on the cells of the mononuclear phagocyte system, and A and B red cell antigens are not fully developed in the fetus or neonate.
Altered expression of ABO antigens in leukemia
The association of weakened expression of A, B and/or H antigens with myeloid malignancies, usually acute myeloid leukemia (AML), is well documented.4 In some cases all red cells show weakness of the A antigen, whereas in others two populations of red cells are clearly apparent. In a patient with acute monoblastic leukemia, initially only 2% were agglutinated with anti-A, but in remission the proportion of agglutinable cells rose to 65% before falling again shortly before death.9 Depending on the method used for detection, between 17 and 55% of patients with myeloid malignancies have lower ABH antigenic expression compared with healthy controls.4 In all cases the changes represent a loss or diminution of antigen strength and never the expression of a new red cell antigen. In some cases loss of A or B was associated with increase in H, in some A or B loss was secondary to loss of H, and in a third group there appeared to be concommitant loss of A/B and H.10 Modifications of ABH antigens may be detected before diagnosis of malignancy, indicating a preleukemic state. Loss of an ABH antigen in a patient with a hematologic disorder is generally prognostic of AML.
Depression of A or B antigens in AML and in preleukemic states is usually associated with a severe reduction in red cell A- or B-transferase activity, but little or no reduction in red cell H-transferase activity.11 Although there may be multiple mechanisms underlying the loss of ABH antigens, DNA methylation is significantly associated with silencing of the ABO transcript in patients with myeloid malignancies and the ABO transcript can be re-expressed in leukemic cell lines by treating with a demethylating agent.12
Lewis antigens
The Lewis antigens, Lea and Leb, are not synthesized by erythroid cells, but become incorporated into the red cell membrane from the plasma. Their synthesis from H antigen and from its precursor is catalyzed by an α1,3/4-fucosyltransferase, the product of FUT3, a gene on chromosome 19. This fucosyltransferase competes with the H-(FUT2), A-, and B-transferases in endodermal tissue for acceptor substrate. The antigens known as Lex and Ley, which are not detected on red cells, are isomers of Lea and Leb, respectively.13
ABH and Lewis antigens on tumors
ABH antigens are often absent from glycoproteins and glycolipids of malignant tissue of the gastrointestinal tract, oral cavity, uterine cervix, lung, prostate, breast and bladder, despite being present in the surrounding epithelium. In many cases loss of ABH antigens preceded formation of distant metastases and hence a poor prognosis.14,15 Loss of A or B antigens from tumor cells may increase their motility and, consequently, their ability to form metastases. In addition, this absence of A or B antigens generally arises from disappearance of A- or B-transferase activity and results in an accumulation of H, Leb, Ley, sialyl-Lea or sialyl Lex. Sialyl-Lea and sialyl Lex are ligands for selectins, and their presence promotes the metastatic process by facilitating interaction with distant organs.14,15
ABO antigen loss results from downregulated transcription of the ABO gene, as no A or B mRNA can be detected in high-grade tumors. At least two different mechanisms may be involved, possibly occurring in tumors derived from different souces: 1) loss of heterozygosity (allelic loss) involving deletions within chromosome 9q34, which contain the ABO locus in addition to tumor suppressor genes; 2) hypermethylation of the CpG island of the ABO promoter region, which down-regulates transcription.16,17
Up-regulation of glycosyltransferases in tumors may result in increased levels of certain carbohydrate structures in the plasma.18 The quantity of circulating sialylated-Lea, otherwise known as the CA 19-9 antigen, is widely used as a marker to support diagnosis of colorectal, pancreatic and gastric cancer, and as an aid to prognosis after potential curative surgery.19,20
Another phenomenon associated with malignancy is the incompatible A antigen occasionally expressed on tumors of group O or B people. About 10% of colonic tumors from group O patients, shown to be homozygous for the O1 allele, express A antigen and contain A-transferase activity.21,22 The molecular basis of this O to A conversion is not known, but alternative splicing of ABO RNA, resulting in loss of exons 5 and 6, would introduce no frameshift or translation-termination codons, but would eliminate the single nucleotide deletion in exon 6 of O1 and the putative gene product would be a truncated glycosyltransferase with a potential for A-transferase activity.14 The higher incidence of gastric and ovarian adenocarcinomas in group A people could be due to a suppression of development of tumors bearing an A antigen by the anti-A naturally present in group O and B, but not A, patients.23
The Rh blood-group system
Rh is the most complex of the human blood-group systems. It has 52 well-defined antigens, the most immunogenic of which is D (RH1). Between 82% and 88% of Caucasians, about 95% of black Africans, and almost 100% of people from the Far East are D-positive. The other main Rh polymorphisms are C/c and E/e, two pairs of antithetical antigens. C has a frequency of 70% and c a frequency of 80% in Europeans. In black Africans the frequency of c is much higher and the frequency of C much lower, whereas in eastern Asia the opposite is the case, with C approaching 100%. In most populations E has a frequency of about 30% and e about 98%.4,5
The antigens of the Rh system are encoded by two genes, RHD and RHCE (reviewed in 24 and 25). They are highly homologous and have very similar genomic organization, each containing 10 coding exons arranged in opposite orientation on chromosome 1 (Fig. 37.2).26 RHCE encodes the C/c and E/e antigens, plus many others such as Cw, Cx and VS. RHD encodes the many epitopes of the D antigen. In Caucasians the D-negative phenotype almost always results from homozygosity for a deletion of RHD, whereas about 66% of D-negative Africans have RHDψ, an inactive RHD gene.27 The products of the Rh genes are polypeptides that are palmitoylated but, unlike most cell surface proteins, not glycosylated. The D and CcEe proteins differ by only 32–35 amino acids, depending on CcEe phenotype. Hydropathy analysis of the amino acid sequences of the Rh proteins together with immunologic evidence suggests that the Rh proteins span the red cell surface membrane 12 times, with internal termini and six extracellular loops (Fig. 37.2).
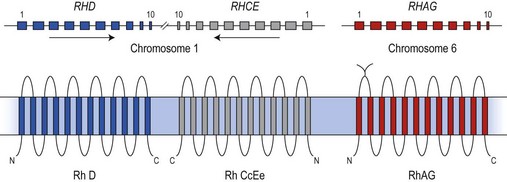
In the D-negative phenotype no D protein is present in the membrane, explaining the high immunogenicity of D compared with other Rh antigens. The C/c and E/e polymorphisms represent amino acid substitutions at different positions in the CcEe protein. Rh epitopes are conformational and may be discontinuous; that is, they are very dependent on the shape of the whole molecule and may involve more than one extracellular loop. Studies with monoclonal antibodies have shown that the D antigen consists of numerous epitopes. There are many variant phenotypes in which some D epitopes are missing, making it possible for a D-like antibody to be produced. Some of these D variants, often referred to as partial D antigens, result from missense mutations in RHD, others because a section of RHD, ranging from part of an exon to several exons, has been replaced by the equivalent region of RHCE. These hybrid RHD-CE-D genes are probably the products of gene misalignment and intergenic recombination during meiosis. RHCE-D-CE hybrid genes are responsible for some CcEe variants.24,25
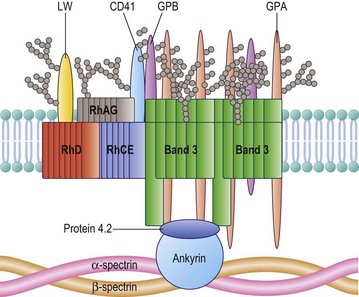
The Rh proteins are closely associated within the membrane with the Rh-associated glycoprotein (RhAG), probably as a heterotrimer.28,29 RhAG has sequence and conformational similarities to the Rh proteins, but has a single N-glycan (Fig. 37.2). This complex is part of a larger protein complex in the red cell membrane, which also includes tetramers of band 3 (the anion transporter, AE1), glycophorins A and B, ICAM-4 (the LW blood group antigen), and CD47, the integrin-associated antigen, in addition to ankyrin and protein 4.2 of the cytoskeleton and some cytosolic proteins, including carbonic anhydrase II and hemoglobin (Fig. 37.3).30 Although the primary function of band 3 is well known – it transfers HCO3− ions across the membrane in exchange for Cl− ions – the function of the Rh protein complex is not known, but there is good evidence that it could act as a gas channel, probably for CO2 and possibly also for O2 and NH3.29–31 The Rh proteins may also be part of another putative membrane complex that includes the glucose transporter (GLUT1), dimers of band 3, glycophorin C, the Duffy (DARC) and Kell glycoproteins, and Xk, which is attached to the cytoskeleton through protein 4.1R, p55, dematin and adducin.32–34
Rh-deficiency syndrome, Rhnull and Rhmod
Red cells with the very rare Rhnull phenotype have no Rh antigens and lack the Rh proteins. The most usual cause of Rhnull is homozygosity for inactivating mutations in RHAG. In the absence of RhAG, no Rh proteins are expressed at the red cell surface. Missense mutations in RHAG may result in reduced Rh antigen expression, a phenotype called Rhmod. Very rarely Rhnull occurs in the presence of RhAG, but with a deletion of RHD and inactivating mutations in RHCE.24,25
Rhnull and Rhmod red cells are morphologically and functionally abnormal. Most Rhnull individuals have some degree of hemolytic anemia associated with stomatocytosis and spherocytosis, which has been referred to as Rh-deficiency syndrome. Rhnull red cells have abnormal organization of their membrane phospholipids, and other anomalies associated with Rhnull include increased cation permeability partially compensated by an increase in the number of K+Na+ pumps, reduced cation and water contents, and significantly reduced CO2 permeability.24,31
HDFN caused by Rh antibodies
The most common form of HDFN results from IgG anti-D crossing the placenta and facilitating the immune destruction of D+ fetal red cells. The severity of anti-D HDFN is highly variable: the most severely affected fetuses die in utero from about the 17th week of gestation onwards; in less severe cases, hydrops fetalis may occur. In those severely affected infants born alive, jaundice may develop rapidly and lead to kernicterus. About 70% of infants who develop kernicterus die within a few days; of those who survive, many have permanent cerebral damage.8
Prior to the late 1960s, HDFN caused by anti-D was a relatively common cause of fetal and neonatal mortality and morbidity. Since then the prevalence of severe anti-D HDFN has been dramatically reduced by anti-D immunoglobulin prophylaxis, in which D− women receive an injection of anti-D IgG within 72 h of delivery of a D+ baby. This prevents immunization of the mother by D+ fetal cells at parturition, protecting D+ fetuses in subsequent pregnancies. In order to reduce the risk of immunization during the pregnancy, it is now policy in some countries to offer one or two doses of anti-D IgG to all D− pregnant women, at around 28–30 weeks’ gestation.35
In Caucasians, only about 60% of fetuses of D− women are D+. When a D− pregnant woman has anti-D, it is beneficial to be able to determine the D phenotype of her fetus in order to determine whether there is any risk from HDFN. Fetal D phenotype can be predicted by polymerase chain reaction-based tests performed on fetal DNA obtained by amniocentesis, chorionic villus sampling or, non-invasively, from cell-free fetal DNA in maternal plasma.36 Basically, the tests determine the presence or absence of RHD in order to predict whether the fetus is D+ or D−, respectively, though modifications are required to prevent errors that would arise from the presence of certain rare variant genes in Caucasians and the relatively common RHDψ in Africans. Where routine antenatal anti-D prophylaxis is offered, it must be offered to all D− pregnant women, as the D type of the fetus is unknown. To prevent unnecessary treatment of pregnant women with blood products, high-throughput technologies suitable for predictng fetal D type from fetal DNA in all D− pregnant women are being developed.37
All antibodies to Rh-system antigens should be considered capable of causing HDFN, but the only Rh antibody other than anti-D that regularly causes severe HDFN is anti-c. Anti-C, -E, -e, and -G have all caused HDFN, but the occurrence is rare and the outcome seldom severe. Methods have been developed for predicting fetal C, c and E from fetal DNA in maternal plasma.36
The Kell system and Xk
The Kell blood group system comprises 32 determinants, almost all of which represent single amino acid substitutions in the Kell glycoprotein.38 The Kell glycoprotein crosses the red cell membrane once, but is unique among blood group proteins as its N-terminal is internal and its large, globular C-terminal domain, which contains six potential N-glycosylation sites and 15 cysteine residues, is external.38 One of the cysteine residues, Cys72, is linked by a disulfide bond to Cys347 of Xk, a multiple membrane spanning protein (see below).39
The Kell glycoprotein is an enzyme; part of the neprilysin family of zinc-dependent endopeptidases. The Kell glycoprotein is able to cleave big endothelin-3, a biologically inactive 40-amino acid peptide, to endothelin-3, a 21-amino acid peptide with vasoconstrictor activity.40 It is not known, however, whether the Kell glycoprotein serves this function in vivo and people who lack the Kell glycoprotein, as a result of inactivating mutations in the KEL gene (Kell-null phenotype), are apparently healthy.
Anti-K and HDFN
Kell-system antibodies can cause severe HDFN. In Caucasian populations anti-K is often the most common immune red cell antibody outside of the ABO and Rh systems.8 Most anti-K appear to be induced by blood transfusion and it is becoming common practice for girls and women of childbearing age to be transfused only with K− red cells.
The pathogenesis of HDFN caused by anti-K differs from that due to anti-D. The severity of the anti-K disease is harder to predict than the anti-D disease. This is because there is very little correlation between anti-K titer and severity of disease and because anti-K HDFN is associated with lower concentrations of amniotic fluid bilirubin than in anti-D HDFN. Postnatal hyperbilirubinemia is not prominent in babies with anemia caused by anti-K. There is also reduced reticulocytosis and erythroblastosis in the anti-K disease, compared with anti-D HDFN. These characteristics suggest that there is less hemolysis in HDFN caused by anti-K, compared with HDFN of comparable severity due to anti-D. This has led to speculation that fetal anemia in anti-K HDFN results predominantly from a suppression of erythropoiesis.41,42 Kell glycoprotein is one of the first erythroid-specific antigens to appear on erythroid progenitors during erythropoiesis, whereas the Rh proteins appear much later.43–45 Vaughan et al46 found that in vitro proliferation of K+ erythroid blast-forming units (BFU-E) and colony-forming units (CFU-E) was specifically inhibited by monoclonal and polyclonal anti-K. They speculated that the Kell glycoprotein might be involved in regulating the growth and differentiation of erythroid progenitors, possibly by enzymatically modulating peptide growth factors on the cell surface. Consequently, binding of anti-K could block the enzymatic activity of the Kell glycoprotein and suppress erythropoiesis. This theory, however, does not take into account the Kell-null phenotype, in which no Kell glycoprotein is present in erythroid cells, yet erythropoiesis is apparently normal. It is likely, therefore, that anti-K suppresses erythropoiesis through the immune destruction of early erythroid progenitors in the fetal liver. Daniels et al.45 have used a functional assay to demonstrate that in the presence of anti-K erythroid progenitors, cultured from cord CD34+ cells derived from a K+ RhD+ baby, elicited a strong response from monocytes; no response was obtained with anti-D because Rh antigens do not appear on erythroid cells before they become hemoglobinized erythroblasts.
The Kell–Xk complex and McLeod syndrome
The 444 amino acid Xk polypeptide is unglycosylated and probably spans the membrane 10 times, with internal N- and C-termini.47 The predicted topographic arrangement is identical to that of members of a family of proteins that cotransport a neurotransmitter together with Na+ and Cl− ions, the amino acid sequence bearing closest resemblance to a Na+-dependent glutamate transporter.47
Xk protein expresses Kx antigen and is covalently linked to the Kell glycoprotein as a disulfide-bonded complex in the red cell membrane.39 The rare absence of Xk gives rise to McLeod syndrome, a multisystem disorder that results from either a gene deletion or from various inactivating mutations within XK, an X-linked gene that encodes the Xk protein.38,47 McLeod syndrome is characterized by weakness of Kell-system antigens and absence of Kx antigen (McLeod phenotype), acanthocytic red cells and elevated serum creatine kinase; late-onset muscular and neurological defects, including muscle wasting, diminished deep tendon reflex, choreiform movements, cardiomyopathy and psychiatric symptoms are common.48 It is likely that absence of Xk from the brain, where it is expressed independently of Kell,49 is responsible for most of the symptoms associated with McLeod syndrome.
A minority of patients with X-linked chronic granulomatous disease (CGD) also have the McLeod phenotype. CGD, an inherited disorder that may be either autosomal or X-linked, impairs the functioning of phagocytes resulting in severe susceptibility to infection. X-linked CGD results from deletion or inactivity of the gene (CYBB) for the beta subunit of flavocytochrome b558, or from mutations within that gene.50 The locus for X-linked CGD and the XK locus are discrete and the association of McLeod phenotype with CGD results from deletions of part of the X-chromosome that encompass both genes.4
Blood groups on red cell transporters
Membrane transporters facilitate the transfer of biologically important molecules in or out of cells. They are typically polytopic, with an even number of α-helical membrane spanning domains of about 21 amino acids each, and have both termini inside the cytosol. Four red cell membrane transporters have blood group activity: band 3, the anion exchanger is the Diego system antigen; aquaporin 1 (AQP1), a water channel, is the Colton antigen; aquaporin 3 (AQP3), a water and glycerol channel, is the Gill antigen; and HUT11, a urea transporter, is the Kidd antigen.3
Band 3 or anion exchanger 1 (AE1), the Diego blood-group antigen, functions as an anion exchanger. It is an antiporter that permits bicarbonate (HCO3−) ions to cross the membrane in exchange for Cl− ions, rapidly reversing the accumulation of HCO3− in the red cells that would occur when CO2 in the blood is hydrated to HCO3− by carbonic anhydrase located in the red cell cytoplasm. This facilitates transport of HCO3− in the plasma, greatly increasing the quantity of CO2 that the blood can convey to the lungs.51
There is only one report of a Diego-null phenotype, a child homozygous for a band 3 mutation, whose red cells had no band 3, and as a consequence no band 4.2, a glycoprotein of the membrane cytoskeleton associated with band 3. The severely hydropic, anemic baby, was delivered by emergency Cesarean section and resuscitated and kept alive by blood transfusion. A cord blood smear revealed dramatic erythroblastosis and poikilocytosis. After 3 years the child was doing reasonably well on a regimen of regular blood transfusions and daily supplements of sodium bicarbonate.52 So, absence of band 3 is compatible with life, but only with extreme medical intervention.
The Kidd red cell glycoprotein is UT-B, a urea transporter,53 which is also present in endothelial cells of the vasa recta, the vascular supply of the renal medulla.54 A urea transporter in red cells has two main functions: 1) transporting urea rapidly in and out the cells to prevent their shrinkage as they pass through the high urea concentration of the renal medulla and subsequent swelling as they leave; and 2) to prevent the red cells from carrying urea away from the renal medulla, which would decrease the urea concentrating efficacy of the kidney.55 Despite this, a Kidd-null phenotype, resulting from homozygosity for a splice site mutation and exon skipping,56,57 is relatively common in Polynesians, with an incidence of about 1 in 400.58 Individuals with the Kidd-null phenotype have no clinical symptoms, but, like UT-B knockout mice, have urine-concentrating ability reduced by about one-third.59,60 The abundance of UT-A and the water channels AQP2 and AQP3 is increased in the renal medulla of UT-B knockout mice, which could assist in the concentration of urea.61 This apparent compensation may explain why Kidd-null individuals have only a modest reduction in urine-concentrating ability. AQP3, but not AQP2, is present on red cells.
Eleven members of the aquaporin family of water channels are found in mammals; two of these, AQP1 (Colton system) and AQP3 (Gill system), are present in human red cells. Aquaporins can be divided into two groups: those like AQP1 that are permeated mostly by water and those like AQP3, known as the aquaglyceroporins, which are permeated by water, but also by other small solutes, especially glycerol.62 In addition to red cells, the highly water permeable AQP1 is present in kidney, lung, vascular endothelium, brain, and eye. AQP1 deficiency may only become important under stress conditions: individuals homozygous for disrupting mutations in AQP1 have the very rare Colton-null phenotype and about 80% reduction in red cell osmotic permeabilities;63 they were apparently healthy, but were unable to concentrate urine maximally when deprived of water.64 In addition to red cells, AQP3 is present in kidney, skin, lung, eye, and colon.62 The very rare Gill-null phenotype, which results from homozygosity for a splice site mutation in AQP3, is not associated with any obvious clinical syndrome.65
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


