When the hemoglobin (Hgb) falls below 4 g/dL the palmar creases lose their pink color, a hallmark of severe anemia.
 Shortness of breath is an underappreciated manifestation of moderate to severe anemia, reflecting insufficient tissue oxygenation.
Shortness of breath is an underappreciated manifestation of moderate to severe anemia, reflecting insufficient tissue oxygenation.
 In organ systems where the circulation is compromised from underlying vascular disease, anemia may precipitate dramatic symptoms, resulting in decompensation of a previously marginally compensated state. This is particularly true for the cerebral and the coronary circulations.
In organ systems where the circulation is compromised from underlying vascular disease, anemia may precipitate dramatic symptoms, resulting in decompensation of a previously marginally compensated state. This is particularly true for the cerebral and the coronary circulations.
Thus in patients with underlying coronary artery disease (CAD), angina or myocardial infarction (MI) may occur, and marginally compensated heart failure may become full blown; more dramatically, in the presence of asymptomatic cerebrovascular disease, a complete hemiparesis may develop which reverts with transfusion and restoration of oxygenation.
Characterization of the Anemia
Red cell indices, developed by Wintrobe in the 1920s, have provided the basis for the classification of anemic states since that time. They provide a measure of the size (MCV) and the Hgb content (MCH and MCHC) of the red cells.
Microcytic Anemias
Hypochromic microcytic anemia (low MCV and low MCH) indicates iron deficiency or hemoglobinopathy; RBCs are pale on smear.
 Anemia with a low MCV in a man means blood loss (iron deficiency) and necessitates a gastrointestinal (GI) workup.
Anemia with a low MCV in a man means blood loss (iron deficiency) and necessitates a gastrointestinal (GI) workup.
An exception is in thalassemia minor where the MCV is very low and the smear is very abnormal.
 Celiac sprue may also cause iron deficiency without bleeding.
Celiac sprue may also cause iron deficiency without bleeding.
 Unusually heavy menses frequently causes iron deficiency in women. The new onset of increased menstrual flow should raise the possibility of thrombocytopenia.
Unusually heavy menses frequently causes iron deficiency in women. The new onset of increased menstrual flow should raise the possibility of thrombocytopenia.
 Hypothyroidism is another cause of heavy menstrual bleeding.
Hypothyroidism is another cause of heavy menstrual bleeding.
 Iron deficiency anemia, especially in women and children, may be associated with pagophagia, a peculiar pica for ice.
Iron deficiency anemia, especially in women and children, may be associated with pagophagia, a peculiar pica for ice.
A history of eating ice cubes should be a tipoff to an underlying iron deficiency anemia. The mechanism is obscure, but the pica resolves after appropriate treatment with iron.
Hemolytic Anemias
Many different processes may result in RBC destruction: mechanical factors, immune-mediated processes, pharmaceuticals, and genetic or acquired alterations in RBC membranes. Evidence of hemolysis includes a rise in indirect bilirubin; a low serum haptoglobin, increased plasma lactic acid dehydrogenase (LDH), and spherocytes on the peripheral smear.
 Rapid turnover of red cell precursors in the marrow of patients with hemolytic anemia may be associated with folate deficiency.
Rapid turnover of red cell precursors in the marrow of patients with hemolytic anemia may be associated with folate deficiency.
Folate replacement, therefore, is part of the treatment of hemolytic anemias.
Microangiopathic Hemolytic Anemia
 Schistocytes and helmet cells indicate a microangiopathic process which has a limited differential and is therefore useful diagnostically. The differential includes thrombotic thrombocytopenic purpura (TTP), disseminated intravascular coagulation (DIC), and malignant hypertension. Malignancy also may cause microangiopathic hemolysis (most commonly with mucinous adenocarcinomas); DIC is the likely cause of the association of microangiopathic hemolytic anemia with cancer.
Schistocytes and helmet cells indicate a microangiopathic process which has a limited differential and is therefore useful diagnostically. The differential includes thrombotic thrombocytopenic purpura (TTP), disseminated intravascular coagulation (DIC), and malignant hypertension. Malignancy also may cause microangiopathic hemolysis (most commonly with mucinous adenocarcinomas); DIC is the likely cause of the association of microangiopathic hemolytic anemia with cancer.
The microangiopathy is caused by activation of the coagulation cascade with fibrin deposition and stranding in small arterioles; these fibrin strands sheer the red cells and damage platelets as they pass through the circulation. In malignant hypertension fibrinoid necrosis in the arterioles is the inciting event that leads to fibrin deposition. Anything that produces widespread endothelial injury may initiate the process including pharmaceuticals, and infectious agents.
 Red cell damage also occurs with dysfunctional cardiac valves, usually artificial or heavily calcified valves, which beat up the red cells (the so-called “Waring Blender” syndrome), thereby causing hemolysis.
Red cell damage also occurs with dysfunctional cardiac valves, usually artificial or heavily calcified valves, which beat up the red cells (the so-called “Waring Blender” syndrome), thereby causing hemolysis.
Autoimmune Hemolytic Anemia
 Autoimmune hemolytic anemia with positive direct Coombs test occurs with lymphoproliferative diseases and with collagen vascular disease, most notably systemic lupus erythematosus (SLE).
Autoimmune hemolytic anemia with positive direct Coombs test occurs with lymphoproliferative diseases and with collagen vascular disease, most notably systemic lupus erythematosus (SLE).
In acquired autoimmune hemolytic anemia the patients’ autoantibodies interact with RBC components; the affected RBCs are cleared in the spleen.
 Since the hemolysis in autoimmune hemolytic anemias occurs principally outside the circulation haptoglobin is reduced only slightly or not at all, despite the fact that the hemolytic process may be quite severe.
Since the hemolysis in autoimmune hemolytic anemias occurs principally outside the circulation haptoglobin is reduced only slightly or not at all, despite the fact that the hemolytic process may be quite severe.
The autoantibodies are said to be “warm” in that hemolysis occurs at normal body temperature (37 °C). These consist primarily of IgG.
 The direct Coombs test detects the autoantibodies on the patients’ RBCs by agglutination of the red cells when incubated with antiglobulin sera raised in animals.
The direct Coombs test detects the autoantibodies on the patients’ RBCs by agglutination of the red cells when incubated with antiglobulin sera raised in animals.
The indirect Coombs test detects antibodies in the patients’ sera directed to normal subjects’ red cells. It is used in type and cross match and in detecting transfusion reactions.
 Hemolytic anemia in conjunction with immune (idiopathic) thrombocytopenia (ITP), known as Evans syndrome, frequently turns out to be SLE in younger patients, and a lymphoproliferative disorder in older folks.
Hemolytic anemia in conjunction with immune (idiopathic) thrombocytopenia (ITP), known as Evans syndrome, frequently turns out to be SLE in younger patients, and a lymphoproliferative disorder in older folks.
Evans syndrome should provoke a workup for lupus and/or lymphocytic malignancy depending on the clinical features.
 Cold agglutinins are antibodies that bind to red cells at temperatures below 37 °C; the resulting hemolysis occurs in cold exposed areas and is usually mild. Mycoplasma pneumonia and infectious mononucleosis are common infections associated with the formation of cold agglutinins.
Cold agglutinins are antibodies that bind to red cells at temperatures below 37 °C; the resulting hemolysis occurs in cold exposed areas and is usually mild. Mycoplasma pneumonia and infectious mononucleosis are common infections associated with the formation of cold agglutinins.
A bedside test showing red cell clumping in an anticoagulated tube of blood when exposed to ice water makes a nice demonstration. The autoantibodies are IgM.
 Hemolytic anemia with leukopenia, thrombocytopenia, and venous thrombosis is highly suggestive of paroxysmal nocturnal hemoglobinuria (PNH).
Hemolytic anemia with leukopenia, thrombocytopenia, and venous thrombosis is highly suggestive of paroxysmal nocturnal hemoglobinuria (PNH).
This rare disorder is caused by acquired hematopoietic stem cell surface protein deficiencies that render the cells sensitive to normal levels of complement in the circulation with resultant intravascular hemolysis and hemoglobinuria.
 Venous thrombosis, particularly of the intra-abdominal vessels including the portal and hepatic veins, is not uncommon in PNH and may reflect a tendency of the platelets to aggregate in the presence of complement.
Venous thrombosis, particularly of the intra-abdominal vessels including the portal and hepatic veins, is not uncommon in PNH and may reflect a tendency of the platelets to aggregate in the presence of complement.
Symptoms are intermittent.
The slight respiratory acidosis that occurs during sleep had been thought to trigger attacks giving rise to the “nocturnal” designation in the name of the disease. This is probably an erroneous explanation. The fact that morning urine is heavily concentrated probably accounts for the red color noted on arising. Hemoglobinuria results from the intravascular hemolysis that occurs in this disease.
The old standby treatments included transfusions, corticosteroids, and supportive care; a newer treatment involves a monoclonal antibody attack on complement.
 Drug-induced hemolytic anemias are most common with penicillin, cephalosporins, methyl dopa, quinine, and sulfonamides.
Drug-induced hemolytic anemias are most common with penicillin, cephalosporins, methyl dopa, quinine, and sulfonamides.
A careful drug history is part of the evaluation of hemolytic anemia.
 Delayed hemolytic transfusion reactions are another cause of hemolysis that is frequently overlooked.
Delayed hemolytic transfusion reactions are another cause of hemolysis that is frequently overlooked.
In persons sensitized by prior blood transfusions or pregnancy an anamnestic antibody response directed against minor RBC antigens (Kidd, Duffy, and Kell, for example) may develop days to weeks after the transfusion and cause hemolysis of the transfused blood. Such reactions are associated with extravascular hemolysis and a positive direct Coombs test which was not present during the pretransfusion cross match because the antibody was present in titers too low to detect.
Megaloblastic Anemias
Megaloblastic anemia results from a defect in red cell DNA synthesis which alters and delays RBC maturation resulting in altered (“megaloblastic”) differentiation. The cause is vitamin B12 or folic acid deficiency; the involvement of B12 results from its role in folate metabolism.
Folate is required for normal DNA synthesis; inadequate availability of folate causes megaloblastic differentiation in the red cell line. The methyltetrahydrofolate trap hypothesis explains how B12 deficiency results in megaloblastosis by reducing folate availability for RBC maturation (Fig. 2-1).
 Vitamin B12, a critical cofactor for methyl group transfers, is required for the regeneration of tetrahydrofolate from methyltetrahydrofolate. Tetrahydrofolate is a form of the vitamin that can be utilized for DNA synthesis. In the absence of B12, folate is trapped in the methylated form which is a metabolic dead end that cannot be utilized for RBC maturation.
Vitamin B12, a critical cofactor for methyl group transfers, is required for the regeneration of tetrahydrofolate from methyltetrahydrofolate. Tetrahydrofolate is a form of the vitamin that can be utilized for DNA synthesis. In the absence of B12, folate is trapped in the methylated form which is a metabolic dead end that cannot be utilized for RBC maturation.
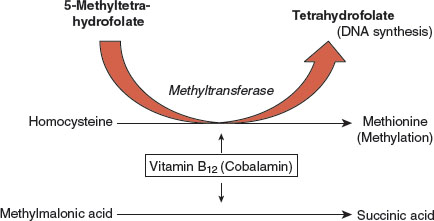
FIGURE 2.1 The methyltetrahydrofolate trap hypothesis: the relationship between folate and vitamin B12 in the pathogenesis of the megaloblastic anemias. Tetrahydrofolate is required for DNA synthesis. In bone marrow rapid cell turnover requires adequate supplies of tetrahydrofolate for normal cell maturation. Methyltetrahydrofolate is a metabolite involved in methyl group transfers including the synthesis of methionine from homocysteine, but is not functional in DNA synthesis. Cyanocobalamin (vitamin B12) is required to regenerate the tetrahydrofolate form which is necessary for normal DNA synthesis and normal red cell maturation. In pernicious anemia (PA) the absence of B12 causes a deficiency of usable folate at the cellular level resulting in megaloblastic differentiation. B12 is also required for myelin synthesis and the conversion of methylmalonic acid to succinate; buildup of methylmalonic acid in the blood is now the preferred test for the diagnosis of PA.
In B12 deficiency, therefore, folate is not available for DNA synthesis, causing a functional folate deficiency at the cellular level and the development of a megaloblastic state.
 The RBCs resulting from megaloblastic differentiation are large (macro-ovalocytes) due to delayed maturation; corresponding changes in the granulocytic cell line lead to multisegmented polymorphonuclear leukocytes.
The RBCs resulting from megaloblastic differentiation are large (macro-ovalocytes) due to delayed maturation; corresponding changes in the granulocytic cell line lead to multisegmented polymorphonuclear leukocytes.
 Nonmegaloblastic macrocytosis may be seen in hypothyroidism, although it should be recognized that hypothyroidism and PA (along with other autoimmune diseases) not infrequently coexist.
Nonmegaloblastic macrocytosis may be seen in hypothyroidism, although it should be recognized that hypothyroidism and PA (along with other autoimmune diseases) not infrequently coexist.
Pernicious Anemia (PA)
Deficiency of vitamin B12 results in PA.
 PA is most common in persons of Northern European descent and is frequently associated with blue eyes, big ears, a sallow yellow complexion, and premature graying of the hair (white before 40 years of age).
PA is most common in persons of Northern European descent and is frequently associated with blue eyes, big ears, a sallow yellow complexion, and premature graying of the hair (white before 40 years of age).
Premature graying is a marker for autoimmune disease in general. The anemia (pallor) in concert with low-grade jaundice may produce a distinctive “lemon yellow” coloration of the skin.
 B12 deficiency is virtually never dietary in origin except in the case of severe and long-standing veganism; B12 deficiency results rather from a failure of the gastric mucosa to produce intrinsic factor which is essential for the normal absorption of the vitamin in the ileum.
B12 deficiency is virtually never dietary in origin except in the case of severe and long-standing veganism; B12 deficiency results rather from a failure of the gastric mucosa to produce intrinsic factor which is essential for the normal absorption of the vitamin in the ileum.
In PA atrophic gastritis is associated with achlorhydria and failure to produce intrinsic factor.
 Ileal disease such as regional enteritis (Crohn’s disease) or surgical ileal resection, total or partial gastrectomy, and blind loop syndromes are other potential causes of vitamin B12 deficiency.
Ileal disease such as regional enteritis (Crohn’s disease) or surgical ileal resection, total or partial gastrectomy, and blind loop syndromes are other potential causes of vitamin B12 deficiency.
 Intestinal motility disorders with bacterial overgrowth in the small bowel may cause B12 deficiency since the bacteria utilize and compete for dietary B12.
Intestinal motility disorders with bacterial overgrowth in the small bowel may cause B12 deficiency since the bacteria utilize and compete for dietary B12.
Folate may be normal under intestinal overgrowth situations as folic acid is synthesized by the overgrown bacteria.
 Since the hepatic stores of B12 are extensive it takes years for the development of anemia to appear after B12 absorption is impaired.
Since the hepatic stores of B12 are extensive it takes years for the development of anemia to appear after B12 absorption is impaired.
The anemia in PA develops chronically and may be very severe with Hgb as low as 3 or 4 g/dL. This necessitates slow and cautious transfusion to avoid pulmonary edema.
 The conversion of methylmalonic acid to succinic acid is a B12 dependent reaction; in B12 deficiency methylmalonic acid builds up and elevated levels have become a standard test for PA (Fig. 2-1).
The conversion of methylmalonic acid to succinic acid is a B12 dependent reaction; in B12 deficiency methylmalonic acid builds up and elevated levels have become a standard test for PA (Fig. 2-1).
Likewise, the conversion of homocysteine to methionine requires B12, so elevated levels of homocysteine are also useful in the diagnosis of PA.
 Plasma levels of B12 levels are much less reliable than methylmalonic levels in the diagnosis of PA.
Plasma levels of B12 levels are much less reliable than methylmalonic levels in the diagnosis of PA.
Autoantibodies may interfere in the B12 assay with erroneous elevation of the B12 level.
 Neurologic changes develop in PA independent of the anemia and consist of demyelination in the lateral and dorsal columns of the spinal cord, resulting in impaired position sense and spasticity of the lower extremities (combined system disease).
Neurologic changes develop in PA independent of the anemia and consist of demyelination in the lateral and dorsal columns of the spinal cord, resulting in impaired position sense and spasticity of the lower extremities (combined system disease).
Clinically, this results in a positive Romberg test and a positive Babinski sign. Personality change and outright dementia may occur as well.
Vitamin B12 is necessary for normal myelin synthesis and deficient supply of this vitamin underlies the neurologic abnormalities that occur in PA. Typical neurologic changes of PA do not occur in folate-related megaloblastic anemias, although peripheral neuropathy may be present with folic acid deficiency.
 Large doses of folic acid may bypass the methyltetrahydrofolate trap, thereby masking the hematologic changes in PA and allowing the serious neurologic consequences of B12 deficiency to progress.
Large doses of folic acid may bypass the methyltetrahydrofolate trap, thereby masking the hematologic changes in PA and allowing the serious neurologic consequences of B12 deficiency to progress.
It is also possible that excess folate obligates the diminished B12 stores to the heme synthesis pathway by drawing B12 from its role in myelin synthesis, thereby worsening the neurologic abnormalities of PA.
 Folic acid supplements should never be given to patients with megaloblastic anemia until PA has been ruled out.
Folic acid supplements should never be given to patients with megaloblastic anemia until PA has been ruled out.
Folate Deficiency
Folic acid deficiency, the other cause of megaloblastic anemia, is caused by inadequate nutrition or impaired folate absorption.
 Folate deficiency frequently occurs in association with alcoholism, and is potentiated by physiologic situations of increased demand, such as pregnancy, and in diseases with increased bone marrow turnover like the hemolytic anemias.
Folate deficiency frequently occurs in association with alcoholism, and is potentiated by physiologic situations of increased demand, such as pregnancy, and in diseases with increased bone marrow turnover like the hemolytic anemias.
 Impaired absorption is also an important cause of folate deficiency. Folic acid in food is conjugated with glutamate and needs to be deconjugated before it can be absorbed.
Impaired absorption is also an important cause of folate deficiency. Folic acid in food is conjugated with glutamate and needs to be deconjugated before it can be absorbed.
 Certain drugs, such as anticonvulsants, estrogens, and alcohol impair the deconjugation of the polyglutamate forms of folic acid that occur naturally in foodstuffs, thus impairing absorption.
Certain drugs, such as anticonvulsants, estrogens, and alcohol impair the deconjugation of the polyglutamate forms of folic acid that occur naturally in foodstuffs, thus impairing absorption.
Folate deficiency is very common in alcoholics due to poor dietary intake and impaired absorption.
 Folate supplements given orally as treatment are not in the polyglutamate form and do not require deconjugation.
Folate supplements given orally as treatment are not in the polyglutamate form and do not require deconjugation.
 Folate malabsorption is common in celiac sprue.
Folate malabsorption is common in celiac sprue.
Hemoglobin Abnormalities
The function of Hgb to bind, store, and release oxygen depends on heme which contains iron in the reduced (ferrous) state.
 The relationship between Hgb and oxygen is expressed in the Hgb–oxygen dissociation curve which represents the percentage saturation of Hgb as a function of the partial pressure of oxygen (Fig. 2-2). Physiologic factors that alter this relationship include pH, temperature, and RBC 2,3 diphosphoglycerate (2,3-DPG, also known as 2,3-BPG).
The relationship between Hgb and oxygen is expressed in the Hgb–oxygen dissociation curve which represents the percentage saturation of Hgb as a function of the partial pressure of oxygen (Fig. 2-2). Physiologic factors that alter this relationship include pH, temperature, and RBC 2,3 diphosphoglycerate (2,3-DPG, also known as 2,3-BPG).
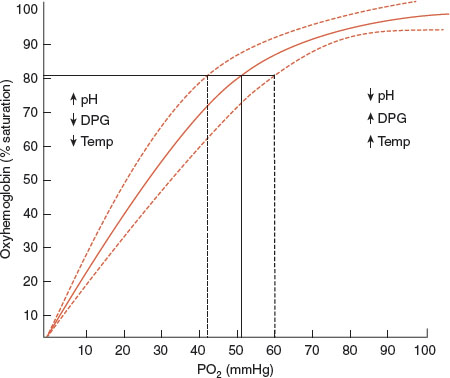
FIGURE 2.2 The oxyhemoglobin dissociation curve: relationship between oxygen saturation of hemoglobin and the partial pressure of oxygen in the blood. Factors that shift the curve to the right (acidosis, fever, and increased 2,3-DPG) enhance oxygen delivery to tissues.
 Acidosis and fever shift the curve to the right and foster enhanced oxygen delivery, while depletion of phosphate, which decreases 2,3-DPG, diminishes the delivery of oxygen and accentuates tissue hypoxia. Several factors related to alterations in the Hgb molecule, such as carbon monoxide exposure which produces carboxyhemoglobin, and methemoglobinemia, also shift the curve to the left, thereby significantly impairing tissue oxygenation.
Acidosis and fever shift the curve to the right and foster enhanced oxygen delivery, while depletion of phosphate, which decreases 2,3-DPG, diminishes the delivery of oxygen and accentuates tissue hypoxia. Several factors related to alterations in the Hgb molecule, such as carbon monoxide exposure which produces carboxyhemoglobin, and methemoglobinemia, also shift the curve to the left, thereby significantly impairing tissue oxygenation.
Oxidative Damage to RBCs
RBCs are particularly vulnerable to oxidative injury since they both carry oxygen and lack cellular organelles and the full biochemical repertory that afford protection to other cells that contain nuclei and mitochondria.
 Oxidative damage to the RBC can occur either by 1) disruption of the cell membrane and denaturation of the RBC proteins which form Heinz bodies or 2) by oxidation of the iron in heme from the ferrous to the ferric form resulting in methhemoglobin.
Oxidative damage to the RBC can occur either by 1) disruption of the cell membrane and denaturation of the RBC proteins which form Heinz bodies or 2) by oxidation of the iron in heme from the ferrous to the ferric form resulting in methhemoglobin.
Distinct clinical syndromes result from each form of damage, and distinct intracellular mechanisms exist to counter the effects of oxidation on RBC proteins and RBC heme respectively.
 An elaborate system protects RBC proteins and membranes from oxidative damage by utilizing the pentose phosphate shunt which generates reduced NADP-H; the latter drives the regeneration of glutathione from its oxidized disulfide state. The glutathione thus formed is the major antioxidant in the red cell which protects RBC proteins and membranes from oxidative damage.
An elaborate system protects RBC proteins and membranes from oxidative damage by utilizing the pentose phosphate shunt which generates reduced NADP-H; the latter drives the regeneration of glutathione from its oxidized disulfide state. The glutathione thus formed is the major antioxidant in the red cell which protects RBC proteins and membranes from oxidative damage.
Insufficient reserves of glutathione results in denaturation of the RBC proteins which clump and attach to the RBC membrane, thus distorting the architecture and damaging the distensibility of the cell. The damaged red cells are removed by the spleen resulting in extravascular hemolysis.
 The clumped denatured RBC proteins are known as Heinz bodies, visible on supra vital staining of the blood smear; the resulting hemolytic anemias are referred to as Heinz body anemias.
The clumped denatured RBC proteins are known as Heinz bodies, visible on supra vital staining of the blood smear; the resulting hemolytic anemias are referred to as Heinz body anemias.
The altered erythrocytes are removed by the spleen (extravascular hemolysis). Removal of the Heinz bodies located at the RBC surface results in characteristic “bite cells.”
The initial step in the reductive resynthesis of glutathione by the pentose phosphate shunt is the oxidation of glucose-6-phosphate catalyzed by the enzyme glucose-6-phosphate dehydrogenase (G6PD).
G6PD Deficiency
 G6PD deficiency, an x-linked recessive trait, and the most common inherited enzyme deficiency in humans, renders the RBCs susceptible to oxidative stress, and the affected patients subject to repeated bouts of acute hemolytic anemia.
G6PD deficiency, an x-linked recessive trait, and the most common inherited enzyme deficiency in humans, renders the RBCs susceptible to oxidative stress, and the affected patients subject to repeated bouts of acute hemolytic anemia.
The disease is most common in patients of Middle Eastern, Mediterranean, and African descent. There is a suggestion, as with sickle cell anemia, that G6PD deficiency may afford protection from malaria, accounting for the persistence of the trait over the course of evolution. Many different mutations have been noted and the severity of the enzyme deficiency depends on the particular mutation. African American males have a mutation associated with more severe disease.
 Many infections, a number of drugs, and certain foods cause oxidative stress and may precipitate hemolytic episodes in G6PD deficient patients.
Many infections, a number of drugs, and certain foods cause oxidative stress and may precipitate hemolytic episodes in G6PD deficient patients.
Although unproven, phagocytosis associated with infections may release oxidants that precipitate attacks. Antimalarials, sulfa drugs, and some antibiotics are the common agents in provoking hemolysis. Fava beans are the classic food that induces attacks. The disease in the Middle East and Mediterranean areas was historically noted as “Favism.”
 The oldest RBCs have the lowest levels of G6PD and are hemolyzed preferentially. This limits the length of each acute episode which ends when the most vulnerable cells have been destroyed.
The oldest RBCs have the lowest levels of G6PD and are hemolyzed preferentially. This limits the length of each acute episode which ends when the most vulnerable cells have been destroyed.
 Assaying for G6PD activity during an acute attack may fail to demonstrate enzyme deficiency since the young RBCs remaining after the older ones are destroyed have sufficient enzymatic activity to yield a false positive result.
Assaying for G6PD activity during an acute attack may fail to demonstrate enzyme deficiency since the young RBCs remaining after the older ones are destroyed have sufficient enzymatic activity to yield a false positive result.
Methemoglobinemia
 Methemoglobin contains iron in the oxidized ferric form rendering it unable to bind oxygen while simultaneously shifting the Hgb–oxygen dissociation curve of the unoxidized ferrous heme to the left. The net result is failure of the RBCs to release oxygen to metabolizing tissues causing tissue hypoxia despite normal PaO2, and turning the color of blood a dark brown.
Methemoglobin contains iron in the oxidized ferric form rendering it unable to bind oxygen while simultaneously shifting the Hgb–oxygen dissociation curve of the unoxidized ferrous heme to the left. The net result is failure of the RBCs to release oxygen to metabolizing tissues causing tissue hypoxia despite normal PaO2, and turning the color of blood a dark brown.
Auto-oxidation of the ferrous ion in heme to the ferric state occurs continuously at a low rate; the ferric ion thus formed is reduced to the ferrous state by an RBC reductase enzyme that utilizes NADH. This limits the level of methemoglobin to less than 1% of the total Hgb concentration. Congenital methemoglobinemia is due to inborn deficiency of the reductase; the more common acquired form is due to drug-induced oxidation of the ferrous ion.
Note that the glutathione reducing system utilizes NADP-H which cannot convert the ferric ion to the ferrous form.
 Local anesthetics, Dapsone, and nitrous oxide are common causes of acquired methemoglobinemia.
Local anesthetics, Dapsone, and nitrous oxide are common causes of acquired methemoglobinemia.
Acute methemoglobinemia during endoscopic procedures may develop as a consequence of using benzocaine or other local anesthetics
 Methylene blue is used therapeutically to reduce the ferric ion in heme to the ferrous state.
Methylene blue is used therapeutically to reduce the ferric ion in heme to the ferrous state.
Under normal physiologic conditions the glutathione reductive pathway cannot reduce the ferric ion to the ferrous state due to the absence, in RBCs, of an electron acceptor for NADPH. Methylene blue provides such an acceptor enabling the glutathione system to reduce the ferric iron and regenerate normal ferrous heme. Note, however, that methylene blue should never be given to patients with G6PD deficiency.
Hemoglobinopathies
Sickle cell anemia, the first disease to be understood in molecular terms, results in clumping of desaturated Hgb, deformation of the affected RBCs, sludging of blood, and occlusion of small vessels resulting in tissue ischemia, manifested clinically as painful crises. Shortened red cell survival leads to anemia. Carriers of the sickle trait (S/A hemoglobin–heterozygote for the S gene) are generally not anemic and do not suffer crises.
 Unexplained bouts of hematuria in young African Americans are usually caused by sickle trait.
Unexplained bouts of hematuria in young African Americans are usually caused by sickle trait.
In individuals with sickle trait RBC sickling does occur in the renal medulla since this area of the kidney is relatively anoxic, and has a high osmolality, both factors which are known to potentiate sickling in S/A RBCs. Arteriolar occlusion is the result with ischemia of the renal medulla leading to papillary necrosis. This usually occurs in adolescence or young adult life, may be clinically silent, or may cause bouts of unexplained hematuria, or renal colic from the sloughed papilla. Isosthenuria with polyuria is the eventual consequence in all patients with sickle trait.
 An abundance of target cells in the peripheral smear is highly suggestive of Hgb C or SC disease.
An abundance of target cells in the peripheral smear is highly suggestive of Hgb C or SC disease.
 Familial cases of polycythemia should raise the suspicion of Hgb variants with increased affinity for oxygen.
Familial cases of polycythemia should raise the suspicion of Hgb variants with increased affinity for oxygen.
Rare Hgb variants have a tighter than normal affinity for oxygen (Hgb –oxygen dissociation curve shifted to the left) with consequent tissue hypoxia and a compensatory increase in RBC production. These unusual variants are a rare cause of polycythemia, frequently occurring in families.
Normochromic–Normocytic Anemia
A wide variety of diseases are associated with normochromic–normocytic red cell morphology.
 Normochromic–normocytic anemia of chronic disease is very common among hospitalized and institutionalized patients; it results from a small increase in red cell destruction and a small decrease in red cell production.
Normochromic–normocytic anemia of chronic disease is very common among hospitalized and institutionalized patients; it results from a small increase in red cell destruction and a small decrease in red cell production.
These changes usually reflect an underlying inflammatory process with associated cytokine production. Diagnostic workup fails to identify a hematologic cause.
 Pure red cell aplasia, aplastic anemia, myelodysplastic syndromes, myelopthisis, and myelofibrosis are all associated with anemia that is usually normochromic–normocytic but may be microcytic.
Pure red cell aplasia, aplastic anemia, myelodysplastic syndromes, myelopthisis, and myelofibrosis are all associated with anemia that is usually normochromic–normocytic but may be microcytic.
The blood smear in these diseases is frequently abnormal with prominent anisocytosis. Examination of the bone marrow is essential for diagnosis and the findings reflect the underlying cause. Frequently the cause is unknown, but in some cases toxin exposure (solvents, particularly benzene), specific infections, or malignancy may be implicated.
PLATELETS
Thrombocytopenia and Purpura
 There are many causes of thrombocytopenia: viral infections, autoimmune destruction, drugs, alcohol, folate deficiency, peripheral consumption (DIC), marrow failure, hypersplenism, cancer chemotherapy, and hematologic malignancies.
There are many causes of thrombocytopenia: viral infections, autoimmune destruction, drugs, alcohol, folate deficiency, peripheral consumption (DIC), marrow failure, hypersplenism, cancer chemotherapy, and hematologic malignancies.
A platelet count below 20,000 μL may be associated with serious bleeding in response to trauma; spontaneous (life-threatening) hemorrhage may occur at counts below 10,000; counts below 50,000 may cause bleeding in response to minor trauma.
 Deficiency of platelets prolongs the bleeding time, tends to be superficial involving the skin and mucous membranes, and occurs promptly after trauma. Menorrhagia is a common manifestation. Petechiae are often the earliest manifestation, coalescing into purpura.
Deficiency of platelets prolongs the bleeding time, tends to be superficial involving the skin and mucous membranes, and occurs promptly after trauma. Menorrhagia is a common manifestation. Petechiae are often the earliest manifestation, coalescing into purpura.
Very low platelet counts may be associated with dangerous central nervous system (CNS) or GI bleeding.
 In distinction to thrombocytopenia, abnormalities of the clotting cascade, such as classic hemophilia, cause deep bleeding into joints (hemarthroses), muscles, and viscera.
In distinction to thrombocytopenia, abnormalities of the clotting cascade, such as classic hemophilia, cause deep bleeding into joints (hemarthroses), muscles, and viscera.
In clotting factor deficiency diseases the whole blood clotting time, and plasma measures of coagulation (partial thromboplastin time [PTT]), are prolonged and bleeding occurs hours after trauma, rather than immediately, since platelets provide the initial hemostatic mechanism.
 Petechiae are small, nonblanching, nonpalpable reddish macules most common on the lower extremities where the hydrostatic forces are greatest. Palpable petechiae occur with vasculitis.
Petechiae are small, nonblanching, nonpalpable reddish macules most common on the lower extremities where the hydrostatic forces are greatest. Palpable petechiae occur with vasculitis.
In contrast, pink morbilliform rashes like those occurring with viral exanthems or drug reactions, blanch with applied pressure.
 A wide variety of common viral infections cause mild thrombocytopenia not associated with bleeding. Usually unnoticed, no treatment or evaluation is necessary. More prolonged or profound thrombocytopenia may occur with human immunodeficiency virus (HIV), hepatitis C virus (HCV), Epstein-Barr virus (EBV), and rickettsial diseases.
A wide variety of common viral infections cause mild thrombocytopenia not associated with bleeding. Usually unnoticed, no treatment or evaluation is necessary. More prolonged or profound thrombocytopenia may occur with human immunodeficiency virus (HIV), hepatitis C virus (HCV), Epstein-Barr virus (EBV), and rickettsial diseases.
The peripheral blood smear must be inspected in every case of thrombocytopenia to ascertain the presence or absence of microangiopathic changes.
 Shistocytes and helmet cells are characteristic of microangiopathy and suggest the diagnosis of TTP.
Shistocytes and helmet cells are characteristic of microangiopathy and suggest the diagnosis of TTP.
The presence or absence of these changes helps identify the underlying cause of the thrombocytopenia and leads to specific therapy: plasmapheresis or intravenous gamma globulin (IVG) for TTP, steroids for ITP.
Idiopathic Thrombocytopenic Purpura (ITP)
The clinical manifestation of ITP is the appearance of petechiae, especially on the lower extremities where they may coalesce into purpuric blotches.
 Also called immune thrombocytopenia, ITP is caused by an autoantibody (IgG) directed against platelet glycoproteins; the tagged platelets are then removed from the circulation by the reticuloendothelial system.
Also called immune thrombocytopenia, ITP is caused by an autoantibody (IgG) directed against platelet glycoproteins; the tagged platelets are then removed from the circulation by the reticuloendothelial system.
 The antiplatelet antibodies in ITP may or may not be detected by available clinical assays, so their absence does not rule out ITP.
The antiplatelet antibodies in ITP may or may not be detected by available clinical assays, so their absence does not rule out ITP.
 The diagnosis of ITP depends on ruling out other causes of thrombocytopenia.
The diagnosis of ITP depends on ruling out other causes of thrombocytopenia.
Since the thrombocytopenia is caused by peripheral destruction, bone marrow aspirate reveals increased megakaryocytes.
 ITP may be triggered by an antecedent infection, especially in children.
ITP may be triggered by an antecedent infection, especially in children.
Prognosis for remission with or without immunosuppression is better in children than in adults.
Thrombotic Thrombocytopenic Purpura (TTP)
 TTP is caused by microvascular thrombi in multiple organs that result in a classic pentad of symptoms and signs: thrombocytopenic purpura, microangiopathic hemolytic anemia, neurologic manifestations, fever, and decreased renal function.
TTP is caused by microvascular thrombi in multiple organs that result in a classic pentad of symptoms and signs: thrombocytopenic purpura, microangiopathic hemolytic anemia, neurologic manifestations, fever, and decreased renal function.
The cause is deficiency of a metalloproteinase that breaks down large aggregates of von Willebrand factor (vWF). The resulting vWF multimers cause platelet clumping and initiate the thrombotic process. The enzyme deficiency may be congenital, but is usually acquired and results from an autoantibody to the enzyme responsible for cleaving the vWF multimers.
Plasmapheresis has dramatically altered the outlook for this disease which was invariably fatal before the benefit of exchange transfusion was recognized.
 Hemolytic-uremic syndrome (HUS) is a related disease mostly of children that usually follows an episode of infectious diarrhea.
Hemolytic-uremic syndrome (HUS) is a related disease mostly of children that usually follows an episode of infectious diarrhea.
Renal failure is more prominent in HUS than in TTP and CNS symptoms less common. Shiga toxin-producing Escherichia coli O157:H7 is the classic, but by no means the exclusive, cause.
Disseminated Intravascular Coagulation (DIC)
 Also known as a consumptive coagulopathy, DIC occurs when the coagulation cascade is inappropriately activated in response to severe infection, malignancy, trauma, toxins, or obstetric emergencies.
Also known as a consumptive coagulopathy, DIC occurs when the coagulation cascade is inappropriately activated in response to severe infection, malignancy, trauma, toxins, or obstetric emergencies.
Uncontrolled intravascular coagulation triggered by exposure of the blood to the procoagulant “tissue factor” and consequent fibrinolysis consume the components of the coagulation system resulting in the paradoxical combination of bleeding and thrombi-induced organ ischemia.
 Thrombocytopenia, prolonged prothrombin time (PT), and prolonged PTT are characteristics of DIC and useful in diagnosis.
Thrombocytopenia, prolonged prothrombin time (PT), and prolonged PTT are characteristics of DIC and useful in diagnosis.
Fibrinogen, which is also consumed in DIC, may or may not be low since it may be elevated as an acute phase reactant in many of the situations in which DIC occurs.
 Fibrin split products (FSPs) and D-dimer are elevated in DIC as a consequence of fibrinolysis.
Fibrin split products (FSPs) and D-dimer are elevated in DIC as a consequence of fibrinolysis.
 Malignancy-associated DIC occurs most commonly with promyelocytic leukemia and mucinous adenocarcinomas.
Malignancy-associated DIC occurs most commonly with promyelocytic leukemia and mucinous adenocarcinomas.
Drug-induced Thrombocytopenias
Aside from chemotherapeutic or immunosuppressive drugs, which affect bone marrow platelet development and have thrombocytopenia as an expected consequence of therapy, many pharmaceuticals cause the peripheral destruction of platelets by an immune mechanism.
 The most common causes of drug-induced thrombocytopenia include heparin, quinidine and quinine, the penicillins, and thiazide diuretics. Of these, heparin is the most common, the most severe, and thus the most important cause.
The most common causes of drug-induced thrombocytopenia include heparin, quinidine and quinine, the penicillins, and thiazide diuretics. Of these, heparin is the most common, the most severe, and thus the most important cause.
There are two types of heparin-induced thrombocytopenia (HIT): Type I, the more common type results from an interaction of heparin with platelets that causes platelet clumping and modest reduction of the platelet count. It is self-limited, begins within 2 days of the initiation of heparin and is not associated with bleeding or thrombotic complications.
 Type II HIT is an immune-mediated reaction in which heparin interacts with platelets, stimulates an antibody against the heparin platelet complex, resulting ultimately in platelet aggregation and endothelial damage. Despite the thrombocytopenia, it is thrombosis, and not hemorrhage, that results and causes the morbidity.
Type II HIT is an immune-mediated reaction in which heparin interacts with platelets, stimulates an antibody against the heparin platelet complex, resulting ultimately in platelet aggregation and endothelial damage. Despite the thrombocytopenia, it is thrombosis, and not hemorrhage, that results and causes the morbidity.
Type II HIT is a potentially severe reaction that typically begins after 4 days of heparin administration. The resulting thrombotic events affect the venous and, less commonly, the arterial circulations. The designation HIT, if unmodified, refers to the type II reaction.
 HIT may be induced by minimal heparin exposure such as that occurring with IV flushes. The reaction is much less common (but does occur) with low molecular weight heparins than with the unfractionated compound.
HIT may be induced by minimal heparin exposure such as that occurring with IV flushes. The reaction is much less common (but does occur) with low molecular weight heparins than with the unfractionated compound.
Treatment, in addition to immediate cessation of all heparin products, involves direct or indirect thrombin inhibitors (not warfarin until the platelet count normalizes). This is essential in all patients since the risk of thrombosis is ongoing for at least 2 to 3 months.
 After an episode of HIT heparin is to be avoided for life.
After an episode of HIT heparin is to be avoided for life.
Other Causes of Petechiae and Purpura
 Petechiae and purpura may also occur with normal platelet counts and normal platelet function. In these cases, vasculitis, capillary fragility, or capillary damage from infection or toxins may be involved.
Petechiae and purpura may also occur with normal platelet counts and normal platelet function. In these cases, vasculitis, capillary fragility, or capillary damage from infection or toxins may be involved.
Henoch–Schönlein purpura, autoerythrocyte sensitization, senile purpura, and scurvy are a few examples.
 Henoch–Schönlein purpura, more common in children but also occurring in adults, is an IgA and complement-mediated vasculitis, in which the petechiae are palpable and the platelet count normal or elevated.
Henoch–Schönlein purpura, more common in children but also occurring in adults, is an IgA and complement-mediated vasculitis, in which the petechiae are palpable and the platelet count normal or elevated.
Arthritis of the large joints and abdominal pain are usually associated.
 Autoerythrocyte sensitization, an obscure and poorly understood entity usually affecting young women, consists of recurrent attacks of painful purpura and ecchymoses involving predominantly the legs. All coagulation studies in these patients are normal.
Autoerythrocyte sensitization, an obscure and poorly understood entity usually affecting young women, consists of recurrent attacks of painful purpura and ecchymoses involving predominantly the legs. All coagulation studies in these patients are normal.
Also known as psychogenic purpura or the Gardner–Diamond syndrome, the attacks may be precipitated by stress and frequently occur in women with emotional problems. The purpura can be reproduced by the subcutaneous injection of RBC stroma.
 Senile purpura, a very common purplish ecchymosis, occurs most prominently on the dorsal forearms of elderly patients. It is caused by loss of subcutaneous connective tissue which leaves the underlying capillaries devoid of support and therefore fragile and liable to bleed in response to minor sheer stress.
Senile purpura, a very common purplish ecchymosis, occurs most prominently on the dorsal forearms of elderly patients. It is caused by loss of subcutaneous connective tissue which leaves the underlying capillaries devoid of support and therefore fragile and liable to bleed in response to minor sheer stress.
No workup is required if the platelet count is normal and the patient is otherwise well.
 In scurvy, the severe vitamin C deficiency results in poor connective tissue support of capillaries leading to a petechial and purpuric eruption.
In scurvy, the severe vitamin C deficiency results in poor connective tissue support of capillaries leading to a petechial and purpuric eruption.
In distinction to other causes of petechiae those associated with scurvy tend to be perifollicular and located on the dorsal, rather than the volar, aspect of the forearm. Large ecchymoses may occur in a saddle distribution involving the buttocks.
 In acute meningococcemia, endotoxin and cytokines damage the endothelium and cause subcutaneous hemorrhage that begins as pink to red petechiae and coalesces to purpura over the course of minutes to hours.
In acute meningococcemia, endotoxin and cytokines damage the endothelium and cause subcutaneous hemorrhage that begins as pink to red petechiae and coalesces to purpura over the course of minutes to hours.
DIC frequently contributes as well.
 Endothelial damage is also the cause of petechiae and purpura in the viral hemorrhagic fevers such as Lassa fever and Ebola infections.
Endothelial damage is also the cause of petechiae and purpura in the viral hemorrhagic fevers such as Lassa fever and Ebola infections.
Again, DIC probably contributes.
OTHER CHANGES IN THE CELLULAR ELEMENTS OF THE BLOOD
Erythrocytosis
 Polycythemia rubra vera, an increase in RBC mass, is a myeloproliferative disease; it needs to be distinguished from “stress polycythemia” in which the RBC mass is normal but the plasma volume is reduced, thereby increasing the concentration of Hgb and the hematocrit.
Polycythemia rubra vera, an increase in RBC mass, is a myeloproliferative disease; it needs to be distinguished from “stress polycythemia” in which the RBC mass is normal but the plasma volume is reduced, thereby increasing the concentration of Hgb and the hematocrit.
Determination of RBC mass with chromium 51-tagged RBCs and consideration of possible other causes will usually clarify the diagnosis. Chronic hypoxia, ectopic production of erythropoietin in paraneoplastic syndromes, and variant Hgbs with tight O2 affinities are other causes of polycythemia.
 Polycythemia vera is usually associated with other abnormalities like splenomegaly, increased uric acid, and physical findings such as ruddy complexion and tortuous and engorged retinal veins (sometimes leading to retinal vein thrombosis).
Polycythemia vera is usually associated with other abnormalities like splenomegaly, increased uric acid, and physical findings such as ruddy complexion and tortuous and engorged retinal veins (sometimes leading to retinal vein thrombosis).
 Hyperviscosity of the blood is noted with hematocrits above 50% and this predisposes to venous thrombosis of the hepatic and other veins.
Hyperviscosity of the blood is noted with hematocrits above 50% and this predisposes to venous thrombosis of the hepatic and other veins.
 Stress polycythemia is due to an increase in catecholamines such as occurs with volume depletion, and with pheochromocytoma. The proximate cause is venoconstriction which initiates diuresis and decreases plasma volume.
Stress polycythemia is due to an increase in catecholamines such as occurs with volume depletion, and with pheochromocytoma. The proximate cause is venoconstriction which initiates diuresis and decreases plasma volume.
The body has no direct means for assessing plasma volume; it uses the surrogate measure of pressure in the capacitance (central venous) portion of the circulation to infer volume status. Venoconstriction with increase in central venous pressure is read as a “full tank” when in fact it is just a smaller tank. Diuresis is thus the consequence of central venoconstriction.
 Acute pancreatitis with exudation of large amounts of fluid in the inflamed abdominal space may be associated with impressive increases in the hematocrit.
Acute pancreatitis with exudation of large amounts of fluid in the inflamed abdominal space may be associated with impressive increases in the hematocrit.
Hematocrits of 60% and above may be seen in severe cases of pancreatitis, and indicates the need for aggressive volume repletion.
Thrombocytosis
 Essential thrombocytosis is part of the spectrum of myeloproliferative diseases; it needs to be distinguished from various other nonmalignant causes of thrombocytosis.
Essential thrombocytosis is part of the spectrum of myeloproliferative diseases; it needs to be distinguished from various other nonmalignant causes of thrombocytosis.
Mild splenomegaly and leukocytosis are frequently associated with essential thrombocytosis. The platelet count exceeds 500,000. The complications are arterial thromboses and paradoxically, bleeding, when the platelet count exceeds one million; bleeding is due to alterations in vWF that occurs in essential thrombocytosis.
 Nonmalignant thrombocytosis occurs with any inflammatory disease, post splenectomy, and with iron deficiency anemia.
Nonmalignant thrombocytosis occurs with any inflammatory disease, post splenectomy, and with iron deficiency anemia.
The elevation is generally modest and thrombotic or bleeding complications do not occur.
 Thrombocytosis in excess of 500,000 may be associated with “pseudohyperkalemia” due to release of potassium from platelets in clotted serum.
Thrombocytosis in excess of 500,000 may be associated with “pseudohyperkalemia” due to release of potassium from platelets in clotted serum.
Measurement of potassium in plasma (as opposed to serum) establishes the true potassium level.
Neutropenias
Neutropenia, also known as granulocytopenia, although the latter technically refers to eosinophils and basophils as well as neutrophils, is defined as an absolute neutrophil count (ANC) below 1,500/μL. Agranulocytosis refers to an ANC of essentially zero.
 Risk of bacterial or fungal infection increases significantly with ANC levels below 500. Viral infections are not impacted since these are defended by lymphocytic immune mechanisms.
Risk of bacterial or fungal infection increases significantly with ANC levels below 500. Viral infections are not impacted since these are defended by lymphocytic immune mechanisms.
Bone marrow evaluation distinguishes between failure of granulocyte development and peripheral destruction. In the presence of neutropenia of uncertain cause the threshold for obtaining a bone marrow aspirate and biopsy should be low.
 Pharmaceuticals, a common cause of neutropenia, may affect neutrophil development in the marrow or cause peripheral destruction by immune mechanisms.
Pharmaceuticals, a common cause of neutropenia, may affect neutrophil development in the marrow or cause peripheral destruction by immune mechanisms.
The list of agents, cancer chemotherapeutics aside, associated with neutropenia is huge. Agranulocytosis is associated with a smaller number of drugs including antithyroid agents, psychotropic agents, and chloramphenicol, the latter causing aplastic anemia as well and no longer in general use.
 The manifestations of agranulocytosis include fever, sores in the mouth and on the gums, sore throat, and trouble swallowing. Patients on agents known to be associated with agranulocytosis need to be warned about the symptoms and instructed to discontinue the drug and call the physician should the above manifestations develop.
The manifestations of agranulocytosis include fever, sores in the mouth and on the gums, sore throat, and trouble swallowing. Patients on agents known to be associated with agranulocytosis need to be warned about the symptoms and instructed to discontinue the drug and call the physician should the above manifestations develop.
This is particularly true for the antithyroid drugs, especially propylthiouracil. These reactions are typically idiosyncratic and the warning is more important than repeated CBCs, although monitoring the latter is recommended as well.
 Infections are common causes of neutropenia. Viral infections and severe bacterial infections are common offenders.
Infections are common causes of neutropenia. Viral infections and severe bacterial infections are common offenders.
 Neutropenia with bacterial sepsis has a poor prognosis.
Neutropenia with bacterial sepsis has a poor prognosis.
Patients with pneumococcal pneumonia are noteworthy examples. Endotoxin from gram-negative organisms, particularly with gram-negative sepsis, is frequently associated with neutropenia. Typhoid fever is a good example where the manifestations of the illness, including neutropenia, reflect endotoxemia.
 Hypersplenism is a prominent cause of pancytopenia affecting RBCs and platelets as well as neutrophils. Any disease associated with splenic enlargement may cause hypersplenism, although congestive splenomegaly from portal hypertension is the most common cause.
Hypersplenism is a prominent cause of pancytopenia affecting RBCs and platelets as well as neutrophils. Any disease associated with splenic enlargement may cause hypersplenism, although congestive splenomegaly from portal hypertension is the most common cause.
Deficiency of any of the formed elements of the blood may occur in hypersplenism, and in any combination.
 Autoimmune neutropenia also occurs in association with several collagen vascular diseases, notably systemic lupus erythematosus and rheumatoid arthritis. Usually modest in degree, the neutropenia is analogous to acquired autoimmune hemolytic anemia and ITP.
Autoimmune neutropenia also occurs in association with several collagen vascular diseases, notably systemic lupus erythematosus and rheumatoid arthritis. Usually modest in degree, the neutropenia is analogous to acquired autoimmune hemolytic anemia and ITP.
In lupus patients under treatment the issue may arise as to the origin of a febrile episode. Is it a lupus flare or infection as a consequence of immunosuppression? In the absence of obvious sepsis, which may lower the WBC count, neutropenia favors a lupus flare.
Lymphopenia and Lymphocytosis
Immunosuppressive drugs such as azathioprine are commonly associated with lymphopenia.
 In the appropriate clinical setting lymphopenia suggests the possibility of HIV infection.
In the appropriate clinical setting lymphopenia suggests the possibility of HIV infection.
 Lymphocytosis is common in several infectious diseases including infectious mononucleosis, cytomegalovirus infections, and importantly, pertussis, where an impressive lymphocytosis may be an important clue to the diagnosis.
Lymphocytosis is common in several infectious diseases including infectious mononucleosis, cytomegalovirus infections, and importantly, pertussis, where an impressive lymphocytosis may be an important clue to the diagnosis.
Leukocytosis
Increases in circulating white blood cells (WBCs) may involve all leukocyte lineages. The particular white cell lines involved has important diagnostic implications.
 Leukemoid reaction refers to neutrophil counts above 25,000 to 50,000, along with the presence of immature neutrophil precursors on blood smear. The usual cause is severe infection.
Leukemoid reaction refers to neutrophil counts above 25,000 to 50,000, along with the presence of immature neutrophil precursors on blood smear. The usual cause is severe infection.
Pneumococcal sepsis is a common cause in young patients and children.
 Leukemoid reactions can be distinguished from chronic myelocytic leukemia (CML) by leukocyte alkaline phosphatase, which is low in CML but elevated in leukemoid reactions.
Leukemoid reactions can be distinguished from chronic myelocytic leukemia (CML) by leukocyte alkaline phosphatase, which is low in CML but elevated in leukemoid reactions.
CML may also be Philadelphia chromosome positive and has an increase in basophils.
 The term leukoerythroblastic reaction refers to nucleated red blood cells along with immature WBCs in the peripheral smear. It occurs with marrow infiltration (myelopthisis), myelofibrosis, or severe infection (sepsis, military TB).
The term leukoerythroblastic reaction refers to nucleated red blood cells along with immature WBCs in the peripheral smear. It occurs with marrow infiltration (myelopthisis), myelofibrosis, or severe infection (sepsis, military TB).
A classic association is with military TB. Eleanor Roosevelt died of military TB with marrow involvement. The leukoerythroblastic reaction had led to the erroneous diagnosis of a hematologic malignancy.
 Eosinophilic leukocytosis occurs with allergic reactions, parasitic infestations (worms, not protozoans), and some collagen vascular diseases (Churg–Strauss syndrome).
Eosinophilic leukocytosis occurs with allergic reactions, parasitic infestations (worms, not protozoans), and some collagen vascular diseases (Churg–Strauss syndrome).
The presence of eosinophils in the blood is a strong evidence against a pyogenic (bacterial) infection.
 Hypereosinophilic syndromes (HESs) are defined as greater than 1,500 eosinophils per μL of blood and accompanied by significant organ infiltration and corresponding organ dysfunction.
Hypereosinophilic syndromes (HESs) are defined as greater than 1,500 eosinophils per μL of blood and accompanied by significant organ infiltration and corresponding organ dysfunction.
HES can be divided into primary, secondary, and idiopathic groups. The primary group is eosinophilia associated with a myeloproliferative disorder, usually a consequence of a chromosomal deletion resulting in constitutive activation of a tyrosine kinase. The abnormality is clonal and sometimes associated with blasts in the peripheral blood. In the secondary group an underlying disorder (parasitic infection, collagen vascular disease, malignancy) produces cytokines that stimulate the eosinophilia. If no cause can be identified the eosinophilia is termed “idiopathic.”
 Pulmonary, cardiac, and gastrointestinal infiltration with eosinophils may be associated with significant morbidity and mortality.
Pulmonary, cardiac, and gastrointestinal infiltration with eosinophils may be associated with significant morbidity and mortality.
Cardiac eosinophilic infiltration is particularly severe affecting both the coronary arteries and the myocardium with corresponding infarction, inflammation, and thrombus formation.
The lungs may be impacted by direct eosinophilic infiltration (chronic eosinophilic pneumonia) or with helminthic larval migration from the intestines to the venous blood and thence to the alveoli and up the respiratory tree to be swallowed and re-enter the gut. The latter, known as Löffler’s syndrome, occurs with ascaris, hookworm, and Strongyloides stercoralis and may be asymptomatic or associated with cough dyspnea and, occasionally, hemoptysis.
 Strongyloides is unique in that the life cycle does not require another host, so autoinfection can occur.
Strongyloides is unique in that the life cycle does not require another host, so autoinfection can occur.
Occasionally, particularly in immunosuppressed patients, a large parasite burden of eggs and larvae can result in strongyloides “hyperinfection,” a serious complication with wide dissemination of the parasite. Strongyloides does occur in the southeastern part of the United States but is more common in immigrants from Southeast Asia.
 GI infiltration with eosinophils may result in abdominal pain, weight loss, vomiting, and diarrhea.
GI infiltration with eosinophils may result in abdominal pain, weight loss, vomiting, and diarrhea.
 Lymphocytic leukocytosis suggests infectious mononucleosis (EBV), pertussis, cytomegalovirus (CMV) infection, or chronic lymphocytic leukemia (CLL). Monocytosis suggests TB.
Lymphocytic leukocytosis suggests infectious mononucleosis (EBV), pertussis, cytomegalovirus (CMV) infection, or chronic lymphocytic leukemia (CLL). Monocytosis suggests TB.
THROMBOTIC DISORDERS AND COAGULOPATHIES
Platelets provide the first line of defense in response to insults that cause bleeding. The subsequent development of a firm clot depends on normal blood coagulation. In addition to the well described inherited clotting factor deficiencies like classic hemophilia, several other acquired coagulopathies cause significant bleeding. At the other end of the spectrum a number of congenital or acquired diseases result in pathologic clotting and thromboembolic disease.
Prothrombotic Diatheses
 Risk factors for the development of deep vein thrombosis (DVT) include stasis in the venous system, blood vessel damage, and hypercoagulability of the blood (Virchow’s triad). Older age independently increases the risk of DVT.
Risk factors for the development of deep vein thrombosis (DVT) include stasis in the venous system, blood vessel damage, and hypercoagulability of the blood (Virchow’s triad). Older age independently increases the risk of DVT.
Thus, immobility, trauma, and factors that exert a prothrombotic effect, such as Factor V Leiden, antiphospholipid antibody syndrome, and estrogens all predispose to DVT and venous thromboembolism (VTE).
 The most significant inherited hypercoagulable state is Factor V Leiden, a mutation that causes resistance of Factor V to inactivation by activated protein C and predisposes to venous thrombosis.
The most significant inherited hypercoagulable state is Factor V Leiden, a mutation that causes resistance of Factor V to inactivation by activated protein C and predisposes to venous thrombosis.
An autosomal dominant trait with variable expressivity Factor V Leiden occurs in the heterozygous state in about 5% of the United States population. The Factor V Leiden mutation is found in about one-quarter of patients presenting with DVT or VTE. Many individuals with the mutation do not experience DVTs. The much rarer homozygous state is associated with a considerably higher incidence of thrombosis.
 The antiphospholipid syndrome (APS) is an autoimmune disease associated with both venous and arterial thromboses.
The antiphospholipid syndrome (APS) is an autoimmune disease associated with both venous and arterial thromboses.
 Pregnancy complications including fetal loss also occur as a clinical manifestation of APS.
Pregnancy complications including fetal loss also occur as a clinical manifestation of APS.
 The autoantibodies are directed against membrane phospholipids and their associated proteins.
The autoantibodies are directed against membrane phospholipids and their associated proteins.
The three antiphospholipid autoantibodies measured clinically are the lupus anticoagulant, the anticardiolipin antibody, and the anti–β2-glycoprotein antibody. (The “lupus anticoagulant” is a misnomer since it is associated with thromboses, not bleeding, and many patients with this antibody do not have lupus. It was called an anticoagulant since it falsely prolongs the PTT.) In addition to detecting these antibodies on two separate occasions the diagnosis of APS requires a clinical event (thrombosis or obstetric) since many normal people have antiphospholipid antibodies without evident disease.
The so-called primary APS occurs in the absence of other rheumatic diseases; secondary APS occurs in conjunction with other autoimmune diseases, most notably, lupus.
 DVT is the most common venous thrombotic event and stroke the most common arterial event in patients having APS. Myocardial infarctions also occur.
DVT is the most common venous thrombotic event and stroke the most common arterial event in patients having APS. Myocardial infarctions also occur.
Perhaps 10% to 15% of these thrombotic events occur in association with APS. It is not clear how these antibodies cause thromboses.
 There is a distinct association of APS with acute adrenal insufficiency.
There is a distinct association of APS with acute adrenal insufficiency.
The adrenal lesion is infarction, either thrombotic or hemorrhagic. The latter may reflect adrenal vein thrombosis with consequent hemorrhagic infarction. Back pain and hypotension are early clues to the diagnosis, and indicate the need for a cosyntropin test.
 Nephrotic syndrome causes a hypercoagulable state by the urinary loss of low molecular weight proteins that block coagulation, particularly, antithrombin III.
Nephrotic syndrome causes a hypercoagulable state by the urinary loss of low molecular weight proteins that block coagulation, particularly, antithrombin III.
Increases in prothrombotic factors, including fibrinogen, also contribute. Perhaps one-quarter of patients with the nephrotic syndrome suffer thrombosis.
 Renal vein thrombosis is particularly associated with the nephrotic syndrome since the deficiency of antithrombin III is greatest in blood draining the kidneys.
Renal vein thrombosis is particularly associated with the nephrotic syndrome since the deficiency of antithrombin III is greatest in blood draining the kidneys.
The procoagulant activity is therefore accentuated in the renal veins increasing the susceptibility to thromboses.
Coagulopathies
 Circulating anticoagulants are autoantibodies that neutralize clotting factors and mimic congenital clotting factor deficiencies except for their initial occurrence in late adult life or after pregnancy. Antibody to Factor VIII is the most common.
Circulating anticoagulants are autoantibodies that neutralize clotting factors and mimic congenital clotting factor deficiencies except for their initial occurrence in late adult life or after pregnancy. Antibody to Factor VIII is the most common.
The diagnosis of a circulating anticoagulant is made by a classic mixing study which tests the result of mixing a patient’s blood with normal plasma. Unlike factor deficiencies the prolongation of the PTT in patients with a circulating anticoagulant is not corrected by a one-to-one dilution with normal plasma. The treatment is with immunosuppressants rather than factor replacement.
 von Willebrand disease (vWD), a congenital deficiency of vWF, produces a mild bleeding diathesis by interfering with normal platelet adhesion at a site of injury.
von Willebrand disease (vWD), a congenital deficiency of vWF, produces a mild bleeding diathesis by interfering with normal platelet adhesion at a site of injury.
The bleeding time is prolonged in vWD. Bruisability, heavy menstrual flow, and bleeding after surgical procedures are common manifestations. Factor VIII is modestly reduced in most cases as well. Replacement of vWF or the administration of DDAVP, which augments the release of vWF, constitutes treatment which may be required before surgical or dental procedures.
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


