Biliary Atresia—Hepatoportoenterostomy
Frederick C. Ryckman
Gregory M. Tiao
Biliary atresia is a serious and potentially fatal liver disease affecting infants. The progressive, sclerosing obliteration of the extrahepatic biliary tree, coupled with progressive inflammatory destruction of the intrahepatic bile ducts, leads to the development of endstage liver disease in the first 6 to 12 months of life. At present, there is no medical therapy to abate or reverse this process. Untreated, the resulting cholestasis leads to progressive conjugated hyperbilirubinemia, cirrhosis, hepatic failure, and death. The hepatoportoenterostomy, introduced by Kasai in 1959, remains the only potentially corrective operative procedure for biliary atresia. The eventual progression to liver transplantation in many children, combined with the reports of increased blood loss and operative complications in the children who have undergone a prior Kasai procedure, has stimulated discussion concerning the use of liver transplantation as primary therapy without prior hepatoportoenterostomy. However, the very acceptable results in selected long-term survivors following the Kasai procedure, contrasted to the risks of liver transplantation and lifelong immunosuppression, support the sequential application of Kasai hepatoportoenterostomy followed, when necessary, by liver transplantation in infants with biliary atresia.
Biliary atresia was first reported in the English language by Burns in 1817, who described the characteristic combination of early jaundice and acholic stools, ascribing this to “some incurable state of the biliary apparatus.” Modern surgical treatment has evolved from the histopathological studies of the progression of biliary atresia by Mario Kasai in the early 1959 when he suggested that the removal of atretic bile ducts before progression of an inflammatory process into the porta hepatis and intrahepatic biliary system might allow successful bile drainage. The hepatoportoenterostomy he described (Kasai procedure) has undergone modifications but remains the template for present surgical therapy.
Etiology
Despite its importance to child health, the etiology of biliary atresia remains unclear. Proposed mechanisms include environmental, autoimmune, and infectious etiologies. Epidemiological studies suggest that ethnic and genetic factors may also play a
role in the pathogenesis of biliary atresia. In the United States, the incidence of biliary atresia is about two times higher in African-Americans as compared with white infants, and it is more common among Chinese than Japanese or Caucasian infants. Girls are more commonly affected.
role in the pathogenesis of biliary atresia. In the United States, the incidence of biliary atresia is about two times higher in African-Americans as compared with white infants, and it is more common among Chinese than Japanese or Caucasian infants. Girls are more commonly affected.
In the 1970s, it was first proposed that a viral agent was the root cause of a group of neonatal obstructive cholangiopathies including biliary atresia. Initial attention focused on the hepatitis B virus. Subsequent studies found several different pathologic viruses in the liver of infants with biliary atresia, including rotavirus, reovirus, cytomegalovirus, human papilloma virus; however, no single virus has been detected uniformly among the patient samples tested. A unifying concept that explains the patient-based results postulates that the clinical manifestations of biliary atresia represent an end phenotype caused by a variety of in utero or perinatal infections. These insults act as a trigger for a host immune response that results in a progressive destruction of the biliary tract. Multiple recent studies support this concept. In a patient-based study, functional genomics was performed on liver biopsy samples obtained at the time of Kasai procedure and microarray analysis revealed coordinated activation of genes involved in lymphocyte differentiation with over-expression of two important Th1 regulatory cytokines—interferon-γ and osteopontin. These cytokines play an important role in the activation of cell-mediated immunity. A subsequent study using RT-PCR and immunohistochemistry confirmed these findings. Although the specific stimulus for the activation of cell-mediated immunity was not identified in either study, cell-mediated immunity is the primary mechanism used by a host to eliminate an infecting virus.
The experimental model of biliary atresia first described in 1993 is important evidence in favor of a viral etiology in the pathogenesis of biliary atresia and has become a vital tool in biliary atresia research. In this model, rhesus rotavirus (RRV) infection of newborn BALB/c mice results in an inflammatory cholangiopathy and pathologic phenotype similar to human biliary atresia including: (a) a clinical progression to obstructive jaundice as manifest by bilirubinuria, jaundice, acholic stools, growth retardation, and eventually death, (b) morphologic and histologic changes in the biliary tree that parallel those found in infants, and (c) a temporal relationship between neonatal exposure to the virus and disease induction. Recent work utilizing this model has linked the pathogenesis of biliary obstruction to the immune effector cells—activated NK and CD8 T-cells. NK cells contribute to the initiation of bile duct injury and CD8 T-cells propagate the injury. Depletion of these cells followed by infection with RRV resulted in virus within the liver but no biliary obstruction. NK cells are an integral component of the innate immune system. CD8+ T-cells are central regulators of the host immune system. Both cell types interact with antigen-presenting cells that have processed pathogen-induced protein accelerating their function. Activation of both NK and CD8 T-cells may play a role in autoimmune disease.
Genetic mechanisms likely play important roles, perhaps influencing susceptibility to other specific causes, but no gene whose altered function would result in atresia of the biliary tree has been identified. The two primary presentations, fetal/embryonic and postnatal, suggest that multiple factors may play roles individually or in combination in specific patients.
Anatomic Classification
The anatomic classification of biliary atresia is descriptive of the degree of extrahepatic ductal destruction. The sclerosing obliterative destructive inflammatory process may affect any length of bile duct from the sphincter of Oddi to the porta hepatis. Type I (10%) demonstrates obliteration of the common bile duct; Type II (2%) also includes common hepatic duct atresia. Both have relative sparing of the intrahepatic ducts and historically have been called “correctable” biliary atresia. Ductal reconstruction using a hepaticojejunostomy yields good results when these less common anatomic variants are diagnosed in infancy. Type III (88%) biliary atresia involves the right/left hepatic ducts and extends into the intrahepatic bile ducts as well.
Clinical Classification
The clinical forms of biliary atresia can be divided into two subtypes: a postnatal form that progresses after birth and a fetal/embryonic form that is present at birth and is often associated with other congenital anomalies.
The postnatal presentation accounts for 65% to 90% of the cases of biliary atresia. These infants present with jaundice which appears after “physiologic jaundice” should have resolved (3 to 4 weeks) and increases in severity within weeks. Hepatic histology is significant for the presence of bile duct remnants with acute and chronic inflammation within the portal triads. This inflammation appears to be progressive. Associated anomalies are uncommon.
The fetal/embryonic subtype occurs in 10% to 35% of infants with biliary atresia, and is characterized by jaundice that is recognized and progresses during the first few days of life. In most cases, no jaundice-free interval can be identified. The probable in utero onset of this is suggested by the absence of patency in the biliary structures and an absence of bile duct remnants at early laparotomy. Associated congenital anomalies are common and follow one of the three patterns: (a) Most common group (60%)— Cardiovascular (Atrioventricular Canal Defects, septal defects, valvular abnormalities, Tetralogy of Fallot, patent ductus arteriosus), Genitourinary (Kidney or collecting system), and Gastrointestinal (Meckel’s diverticulum, Midgut anomalies), (b) Laterality Sequence anomalies (30%)—polysplenia, asplenia, situs inversus, intestinal malrotation, cardiovascular anomalies (BASM), and (c) Isolated intestinal malrotation (10%).
Recent review of the U.S. and U.K. experience has confirmed the worse overall prognosis for infants with the BASM phenotype. Most studies have demonstrated a poorer prognosis with any fetal/embryonic subtype.
Table 1 Evaluation of Infants with Cholestasis | ||
|---|---|---|
|
Clinical Course
To be successful, the Kasai procedure should be done before irreversible sclerosis of the intrahepatic bile ducts has occurred. Prompt evaluation is necessary for any infant older than 14 days with jaundice to determine if conjugated hyperbilirubinemia is present (Table 1). If infectious, metabolic,
and endocrine disorders are excluded, then hepatic biopsy most accurately identifies infants with suspected biliary atresia. In these selected infants, exploratory laparotomy and intraoperative examination/cholangiogram should be done expeditiously by a surgeon who has experience undertaking the Kasai procedure.
and endocrine disorders are excluded, then hepatic biopsy most accurately identifies infants with suspected biliary atresia. In these selected infants, exploratory laparotomy and intraoperative examination/cholangiogram should be done expeditiously by a surgeon who has experience undertaking the Kasai procedure.
Preliminary studies to identify biliary atresia are rarely definitive. Although biochemical evidence of cholestasis and hepatocellular damage is present, the biochemical tests are of no discriminating value in patients with suspected biliary atresia. Abdominal ultrasonography excludes choledochal cyst, perforation of the bile ducts, or choledocholithiasis. In patients with suspected biliary atresia, the gall bladder is either not visualized or atretic. When a hyperechogenic liver hilum is identified, known as a “triangular cord” sign, biliary atresia is likely. Associated anomalies such as a pre-duodenal portal vein, polysplenia/asplenia, inferior vena caval interruption, or malrotation can also be identified. These findings are helpful to the surgeon and suggest biliary atresia but are not diagnostic of the disorder. Cholescintigraphy is rarely helpful even with phenobarbital induction, and often delays critical surgical intervention.
There is no clinical, laboratory, or histologic factor which has been reliably predictive of success following hepatoportoenterostomy. Factors recognized to affect prognosis in biliary atresia are listed in Table 2. The patient’s age at operation was shown to directly affect prognosis in Kasai’s original series. However, the significance of age at operation is inconsistent. The French National study reported decreased 5 year survival with the native liver in patients >90 days of age at operation. Conversely, Davenport et al. in the King’s College, London, series showed no significant difference in clearance of jaundice or 2 year native liver survival with increasing age in isolated biliary atresia. Patients with BASM anomalies showed worse outcomes with advancing age.
Histologic examination of the liver is the most helpful step in evaluating the children with suspected biliary atresia and selecting appropriate candidates for laparotomy. The histopathologic findings that predict obstructive forms of neonatal cholestasis are portal ductal proliferation, bile plugs in portal bile ductules, portal-portal bridging, neutrophils, hepatocyte swelling, and multinucleated giant hepatocytes. In preoperative liver biopsies, the presence of giant cells, lobular necrosis, focal necrosis, bridging necrosis, and cholangitis are associated with poor clinical outcome, whereas the presence of bile in zone 1 is associated with improved clinical success. Success in achieving bile drainage correlated directly with the size of the bile duct remnants identified in the porta hepatis at surgery—bile duct profiles >150 u have the best prognosis and ductal remnants <50 u in size uniformly fair poorly. The degree of portal fibrosis is especially important in infants who present at a later age for evaluation. The combination of late presentation (>3 months of age) and severe fibrosis makes successful bile drainage unlikely; in these cases, primary liver transplantation should be considered as the preferred option. The steps for rapid evaluation of the infant with suspected biliary atresia are outlined in Table 3.
Table 2 Factors Affecting Prognosis in Biliary Atresia | |
|---|---|
|
Table 3 Recommended Steps in the Evaluation of a Child with Possible Biliary Atresia | |
|---|---|
|
A favorable impact from regionalization of care for biliary atresia has been demonstrated in several studies. In the United Kingdom, biliary atresia surgery was restricted to three centers in 1999 when significant patient survival and native liver survival differences were noted in centers with <5 cases/year operative volume. Regionalization improved the overall outcomes. The French National Study (1986 to 1996) also noted significantly improved native liver, post-liver transplant and overall patient survival in a high volume (>20/y) center compared to the ones treating <5 cases/year. In both studies, surgeon experience with the Kasai Portoenterostomy and overall center experience with liver disease and liver transplantation were felt to be critical components. In the USA, success rates similar to those in the UK and France have been recorded by the Biliary Atresia Research Consortium (BARC), representing nine clinical centers with established liver care centers. This strongly suggests that biliary atresia patients achieve the best outcomes in the centers focused on their specific disease type and surgical needs.
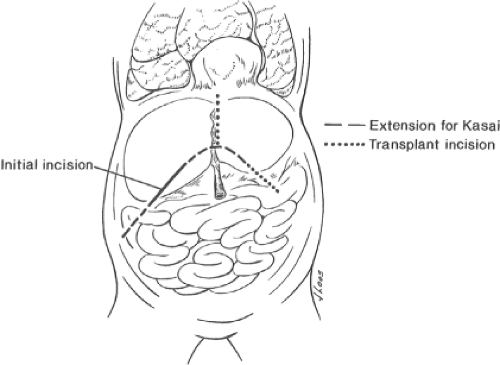 Fig. 1. The initial incision for exploration is a right subcostal approach. If portoenterostomy is indicated, this is extended in the medial direction slightly across the midline and also to the right. This incision can later be converted to a transplant approach if necessary.
Stay updated, free articles. Join our Telegram channel
Full access? Get Clinical Tree
 Get Clinical Tree app for offline access
Get Clinical Tree app for offline access

|