241. A 3-year-old boy immigrant from Somalia presents for evaluation of priapism (prolonged and painful erection). The parents report through a translator that he has an illness well known in their family, and that several cousins are affected. The illness causes fatigue, poor growth, and bone pain, and the physician suspects sickle cell anemia (MIM*603903). Sickle red cells are observed on peripheral smear, and the physician suggests confirmation by DNA diagnosis so the couple can consider preimplantation/prenatal diagnosis with future pregnancies.
The β-globin gene is diagrammed in the upper portion (A) of the figure, consisting of untranslated RNA regions (gray boxes), three exons (clear boxes), and two introns (lines between exons). DNA diagnosis for sickle cell anemia was facilitated by finding that a cleavage site for restriction endonuclease (MstII) included the sixth codon of β-globin gene exon 1 (solid circle in first exon). When this codon is mutated to cause the glutamate to valine amino acid change in sickle cell anemia, the MstII site is ablated. MstII sites in normal β-globin genes are shown as solid arrows in the upper part of the figure, yielding gene fragments of 165 bp (5′ MstII site to that in the glutamate codon) and 515 bp (MstII site in glutamate codon to the 3′ MstII site). Recall that the β-globin gene is on chromosome 16, and that both copies are mutated (homozygous) in individuals with sickle cell anemia. Analysis of β-globin gene structure and expression in an individual with sickle cell anemia would yield which of the following results? (Southern blotting is performed using MstII endonuclease cleavage and hybridization with the β-globin gene probe shown in the figure.)
a. MstII DNA cleavage segment of 515 and 165 bp by Southern blot, nucleic acid segment of ∼550 bp by Northern blot, and abnormal protein by hemoglobin electrophoresis
b. MstII DNA cleavage segments of 515 and 165 bp by Southern blot, no nucleic acid segment detected by Northern blot, and abnormal protein by hemoglobin electrophoresis
c. MstII DNA cleavage segments of 515 and 165 bp by Southern blot, nucleic acid segment of ∼1400 bp by Northern blot, and abnormal protein by hemoglobin electrophoresis
d. MstII DNA cleavage segment of 680 bp by Southern blot, nucleic acid segment of ∼550 bp by Northern blot, and abnormal protein by hemoglobin electrophoresis
e. MstII DNA cleavage segments of 680, 515, and 165 bp by Southern blot, nucleic acid segment of ∼550 bp by Northern blot, and abnormal protein by hemoglobin electrophoresis
242. An 18-month-old Caucasian boy of Italian ancestry is evaluated for “failure to thrive”—his height and weight have leveled off and have gone from the 50th to the 3rd percentiles for age. Parental neglect is suspected and he is hospitalized for calorie counts and daily weights to see if normal feeding results in weight gain. Upon physical admission, a medical student notes he is pale with brownish-yellow pigmentation to his skin, and that his face seems unusual. Because of these observations, laboratory testing is initiated before the dietary treatment is completed and reveals a low hemoglobin (5 g/dL with normals of 12-14 for age) and target cells on smear that suggest a diagnosis of β-thalassemia (MIM*141900). Molecular studies show a mutation just upstream (5′) to the transcription initiation site of both β-globin alleles (point a in the figure above Questions 241 to 248). In homozygous individuals, this mutation decreased the amount of β-globin mRNA and subsequent β-globin protein and hemoglobin, producing anemia. Which of the following best describes this mutation?
a. It affects a promoter sequence that codes for RNA polymerase, lowering β-globin mRNA levels.
b. It affects a promoter sequence and the rate at which RNA polymerase II initiates transcription, lowering β-globin mRNA levels.
c. It affects the termination site of an upstream gene, increasing β-globin mRNA levels.
d. It alters a sequence encoding a subunit of RNA polymerase II (the sigma factor), increasing α-globin mRNA levels.
e. It affects a promoter sequence and the rate at which RNA polymerase II terminates transcription, increasing α-globin mRNA levels.
243. A 24-year-old African American mountain climber in excellent physical condition suffers shortness of breath and low oxygen (hypoxia) at high altitude in Nepal. After transport to base camp and oxygen treatment, a family history reveals that his mother has sickle cell anemia (MIM*603903). With reference to the upper portion (A) of the figure above Questions 241 to 248, laboratory studies of his β-globin gene structure and expression would be expected to show which of the following results? (Note that the same MstII restriction and β-globin probe in the figure is used for Southern blotting.)
a. MstII DNA cleavage segment of 515 and 165 bp by Southern blot, RNA segment of ∼550 and 700 bp by Northern blot, and normal and abnormal proteins by hemoglobin electrophoresis
b. MstII DNA cleavage segments of 680, 515, and 165 bp by Southern blot, RNA segment of ∼550 and 700 bp by Northern blot, and normal and abnormal proteins by hemoglobin electrophoresis
c. MstII DNA cleavage segments of 515 and 165 bp by Southern blot, RNA segment of ∼1400 bp by Northern blot, and single abnormal protein by hemoglobin electrophoresis
d. MstII DNA cleavage segment of 680 bp by Southern blot, RNA segment of ∼700 bp by Northern blot, single normal protein band by hemoglobin electrophoresis
e. MstII DNA cleavage segments of 680, 515, and 165 bp by Southern blot, RNA segment of ∼1400 and 700 bp by Northern blot, and single abnormal protein by hemoglobin electrophoresis
244. An 8-year-old African American girl presents with severe anemia, despite several years of treatment with iron supplementation. A blood smear is examined carefully and reveals both target cells suggestive of β-thalassemia (MIM*141900) and sickle cells suggestive of sickle cell anemia (MIM*603903). Referring again to the figure above Questions 241 to 248, molecular analysis demonstrates a mutation at the promoter site (point a in the figure) on one β-globin gene and a sickle cell mutation on the other. Which of the following laboratory results would be expected in such an individual, using the same MstII restriction and β-globin probe for Southern blotting described previously?
a. MstII DNA cleavage segment of 515 and 165 bp by Southern blot, normal amounts of ∼700 bp RNA segment by Northern blot, and normal and abnormal proteins by hemoglobin electrophoresis
b. MstII DNA cleavage segments of 680, 515, and 165 bp by Southern blot, decreased amounts of ∼550 and 700 bp RNA segment by Northern blot, and normal and abnormal proteins by hemoglobin electrophoresis
c. MstII DNA cleavage segments of 680, 515, and 165 bp by Southern blot, decreased amounts of a ∼550 and 700 bp RNA segment by Northern blot, and mostly abnormal proteins by hemoglobin electrophoresis
d. MstII DNA cleavage segment of 680 bp by Southern blot, normal amounts of a ∼550 and 700 bp RNA segment by Northern blot, and mostly normal proteins by hemoglobin electrophoresis
e. MstII DNA cleavage segments of 680, 515, and 165 bp by Southern blot, lower amounts of 550 and 700 bp DNA segment by Northern blot, and mostly normal proteins by hemoglobin electrophoresis
245. A population study in a rural area of Greece examined several patients with anemia and revealed a homozygous mutation in a sequence 5′-TATAAAA-3′ at the 5′ end of the β-globin gene (site a in the figure above Questions 241 to 248. This sequence has been found at the 5′ boundary of other eukaryotic genes, and is quite similar to a consensus sequence observed in prokaryotes. Which of the following best describes the significance of this mutation?
a. Likely β-thalassemia due to disruption of RNA polymerase III binding
b. Likely hemoglobinopathy such as sickle cell anemia due to promoter disruption
c. Likely β-thalassemia due to disruption of transcription termination
d. Major binding site of RNA polymerase I
e. Likely β-thalassemia due to disruption of transcription initiation by RNA polymerase II
246. A 2-year-old Caucasian from a Greek community in Chicago presents with severe anemia, targeted red blood cells on peripheral smear, skin pallor, jaundice, and growth failure. Molecular analysis demonstrates a homozygous mutation in the β-globin gene at the junction of exon 1 and intron 1 (site c in the figure above Questions 241 to 248). Which of the following best describes the nature and clinical consequence of this mutation?
a. Altered splice donor site, absent mRNA by Northern blotting, and β-globin protein deficiency presenting as α-thalassemia
b. Altered splice acceptor site, altered mRNA size by Northern blotting, and β-globin protein deficiency presenting as α-thalassemia
c. Altered splice donor site, altered mRNA size by Northern blotting, and β-globin protein deficiency presenting as β-thalassemia
d. Altered promoter site, altered mRNA size by Northern blotting, and β-globin protein deficiency presenting as β-thalassemia
e. Altered promoter site, deficient β-globin mRNA by Northern blotting, β-globin protein deficiency presenting as α-thalassemia
247. A 22-year-old male is found to have a hemoglobin of 11.5 during his army induction physical and his red cell indices include a mean corpuscular volume (MCV) of 79 (mean 90 femtoliters, 80 is –2 SD). The microcytosis and anemia first suggest iron deficiency, but ferritin levels (reflection of total body iron) are normal. A diagnosis of hemoglobin H disease (MIM*141800) is considered, recalling that the α-globin genes are similar in structure to the β-globin genes diagrammed in the figure above Questions 241 to 248, but are present in duplicate copies on each chromosome 16 in humans. Which of the following α-globin mutations would be compatible with moderate anemia of the type seen in hemoglobin H disease α-globin genes?
a. Heterozygous point mutation within the TATA box of one α-globin gene
b. Homozygous point mutation within the TATA box of one α-globin gene
c. Homozygous mutation deleting both α-globin gene copies
d. Heterozygous mutation deleting both α-globin gene copies paired with a heterozygous mutation with the TATA box of one α-globin gene
e. Homozygous frameshift mutation within the coding sequence of the 5′ α-globin gene
248. A population survey of a Northern Italian population reveals a variety of mutations in the β-globin gene. It is known that eucaryotic mRNAs undergo several forms of posttranscriptional processing, and that some forms of thalassemia are due to incorrect processing of the α- and β-globin mRNAs. Referring to the β-globin gene structure shown in the figure above Questions 241 to 248, which of the following homozygous mutations would most likely present as altered hemoglobin (hemoglobinopathy) but not as a β-thalassemia?
a. Mutations changing the consensus AGGUAAGU splice donor sequence at exonintron junctions
b. Missense mutations in exon 2
c. Mutations changing the AAUAA recognition sequence at the terminus (3′-end) of the gene
d. Mutations altering TATA or CAAT boxes
e. Mutations changing the consensus UACUAAC-30bp-CAGG splice acceptor sequence at intron-exon junctions
249. An 18-year-old Ashkenazi Jewish female had developed worsening balance and coordination (ataxia) with declining vision at night. She also had chronic diarrhea that had been attributed to irritable bowel syndrome but was recognized as fat malabsorption from gastroenterology studies. A diagnosis of autosomal recessive abetalipoproteinemia (MIM*200100) was considered and analysis of apolipoprotein B (apoB) gene expression was initiated. Northern blots of the patient’s duodenal biopsy tissue showed an apoB mRNA of expected size, while Western blots demonstrated an apoB peptide that was smaller than that characterized in control liver tissue. A nonsense, chain-terminating mutation was thought likely until the same small peptide was found in intestinal biopsies of controls. Which of the following interpretations is most likely?
a. ApoB mRNA undergoes alternative splicing in intestine; the patient has a heterozygous apoB gene deletion.
b. An exon of the apoB gene is deleted in intestine; the patient has homozygous apoB gene deletion.
c. Intestinal apoB mRNA undergoes a codon alteration that causes termination of translation and a smaller peptide; the patient may have homozygous apoB missense mutation.
d. Polyadenylation of apoB mRNA is deficient in intestine; the patient has heterozygous apoB mutation affecting transcription termination.
e. Transcription of apoB mRNA occurs from a different promoter in intestine; the patient has heterozygous apoB mutation deleting this promoter.
250. A 21-year-old Hispanic female presents for evaluation of recurring kidney stones, leg pain, and blanching of her hands with the cold (Raynaud phenomenon). Her prior care was in Mexico and records of prior stone episodes (urolithiasis) or the nature of the stones are not available. Laboratory testing shows elevated blood urea nitrogen (BUN) and creatinine suggesting chronic renal disease, and urinary excretion of oxalic acid is greatly elevated. A diagnosis of primary oxalosis or hyperoxaluria (MIM*259900) is suggested, known to be caused by abnormal location of alanine-glyoxylate aminotransferase enzyme in the endoplasmic reticulum (ER) rather than the peroxisome. Patients can be treated by liver transplant to restore a source of peroxisomal enzyme, but a less invasive cure could be achieved by engineering one mutant allele so its product enzyme was not targeted to the ER. This alteration would be achieved by which of the following strategies?
a. Cleaving the enzyme’s carboxy-terminal segment
b. Changing RNA splicing to include an extra exon in the mRNA
c. Adding a proteolytic cleavage site near the protein terminus
d. Changing the enzyme’s protein processing to produce a smaller peptide
e. Altering the enzyme’s amino-terminal sequence
251. A 72-year-old Caucasian male experiences progressive shortness of breath, and is diagnosed with emphysema. He had smoked up to one pack per day of cigarettes until 5 years ago. Family history reveals that his father, two paternal uncles, and his father’s mother all died of emphysema, and a diagnosis of α1-antitrypsin deficiency (AAT-MIM*107400) is suspected. He is found to have decreased amounts and abnormal mobility of AAT protein in his serum when analyzed by serum protein electrophoresis. Liver biopsy discloses mild scarring (cirrhosis) and demonstrates microscopic inclusions due to an engorged endoplasmic reticulum. Which of the following is the most likely explanation for these findings?
a. Defective transport from hepatic ER to the plasma
b. A mutation affecting the N-terminal methionine and blocking initiation of protein synthesis
c. A mutation affecting the signal sequence
d. Defective structure of the signal recognition particles
e. Defective energy metabolism causing deficiency of GTP
252. A 6-month-old Caucasian boy has had chronic yeast diaper rash and four episodes of pneumonia, two with positive blood cultures for Group A streptococcus. Knowing that Group A strep infections are rare in infants and that the patient likely has defective B-cells that fight bacteria and defective T-cells that fight viruses and fungi, his physician thinks a diagnosis of severe combined immune deficiency (SCID) due to adenosine deaminase (ADA) deficiency (MIM*102700) is likely. Which of the following treatment options would permanently cure the patient?
a. Germ-line gene therapy to replace one ADA gene copy
b. Germ-line gene therapy to replace both ADA gene copies
c. Somatic cell gene therapy to replace one ADA gene copy in circulating lymphocytes
d. Somatic cell gene therapy to replace both ADA gene copies in circulating lymphocytes
e. Somatic cell gene therapy to replace one ADA gene copy in bone marrow lymphoblasts
253. A 5-year-old Egyptian boy receives a sulfonamide antibiotic as prophylaxis for recurrent urinary tract infections. Although he was previously healthy and well nourished, he becomes progressively ill and presents to your office with pallor and irritability. A blood count shows that he is severely anemic with jaundice due to hemolysis of red blood cells. Which of the following is the simplest test for diagnosis?
a. Northern blotting of red blood cell mRNA
b. Enzyme assay of red blood cell hemolysate
c. Western blotting of red blood cell hemolysates
d. Amplification of red blood cell DNA and hybridization with allele-specific oligonucleotides (PCR-ASOs)
e. Southern blot analysis for gene deletions
254. A 28-year-old female presents for evaluation after failing to become pregnant after 3 years of marriage. Her husband’s sperm counts and her uterine sonogram plus clotting and hormone studies are all normal, and tubal insufflation has failed to influence fertility. Her physician then notes a positive urine reducing substance that turns out to be galactose, and orders testing of the GALT enzyme that converts UDP-galactose (substrate) to UDP-glucose (product) and is responsible for classical galactosemia (MIM*230400). The result shows a slightly low value for GALT activity with none of the common mutant alleles detected by DNA sequencing. The physician recalls the occurrence of ovarian dysfunction in galactosemia and invites collaboration with the pathologist to measure the Km of the female’s GALT enzyme to see if an alteration might justify detailed DNA sequencing studies. The amount of UDP-galactose to UDP-glucose conversion in 1 minute is measured using the same amount of enzyme (E) at 6 increasing UDP-galactose concentrations. Which of the following analyses would be most useful for determining the Km?
a. Plot of E against S
b. Plot of 1/Vi versus 1/S
c. Plot of Vi versus S
d. Plot of 1/E versus 1/S
e. Plot of Vmax versus 1/S
255. A 3-day-old Caucasian girl has a cleft palate, heart defect, extra fifth fingers, scalp defect (absent skin exposing underlying flesh), and unusual face with narrow distance between the eyes. Her physician orders a routine karyotype that shows 46 chromosomes with extra material on one homolog of the chromosome 5 pair. This chromosomal abnormality is best described by which of the following terms?
a. Polyploidy
b. Balanced rearrangement
c. Ring formation
d. Mosaicism
e. Unbalanced rearrangement
256. A 10-year-old African American boy is referred to the physician because of learning problems and some behavior changes. His family history is unremarkable. Physical examination reveals tall stature with few anomalies except for single palmar creases of the hands and curved fifth fingers (clinodactyly). The physician decides to order a karyotype. Which of the following indications for obtaining a karyotype best explains the physician’s decision in this case?
a. A couple with multiple miscarriages or a person who is at risk for an inherited chromosome rearrangement
b. A child with ambiguous genitalia who needs genetic sex assignment
c. A child with an appearance suggestive of Down syndrome or other chromosomal disorder
d. A child with mental retardation and/or multiple congenital anomalies
e. A child who is at risk for cancer
257. A 4-year-old Caucasian girl presents with short stature, web neck, and other features suggestive of Turner syndrome, but also has mild mental disability. Her chromosome studies reveal 90,XX/92,XXXX with about 10% abnormal cells in blood and 20% in skin. These results can be described as which of the following?
a. Aneuploidy
b. Haploidy
c. Triploidy mosaicism
d. Tetraploidy without mosaicism
e. Trisomy with mosaicism
258. Which of the following is the proper cytogenetic notation for a female with Down syndrome mosaicism?
a. 46,XX,+21/46,XY
b. 47,XY,+21
c. 47,XXX/46,XX
d. 47,XX,+21/46,XX
e. 47,XX,+21(46,XX)
259. Children with type IV glycogen storage disease (MIM*232500) accumulate abnormal glycogen, causing them to have progressive liver damage in addition to the low blood glucose (hypoglycemia), increased triglycerides and cholesterol (hyperlipidemia), and high uric acid (hyperuricemic) due to deficient glycogenolysis (see High-Yield Facts, Table 3). Type IV glycogen storage is autosomal recessive. Autosomal recessive conditions are correctly characterized by which of the following statements?
a. They are often associated with deficient enzyme activity.
b. Both alleles contain the same mutation.
c. They are more variable than autosomal dominant conditions.
d. Most persons do not carry any abnormal recessive genes.
e. Affected individuals are likely to have affected offspring.
260. A family with retinitis pigmentosa is encountered, and the pedigree shown below is documented. What is the risk that a son born to individual III-3 would be affected?
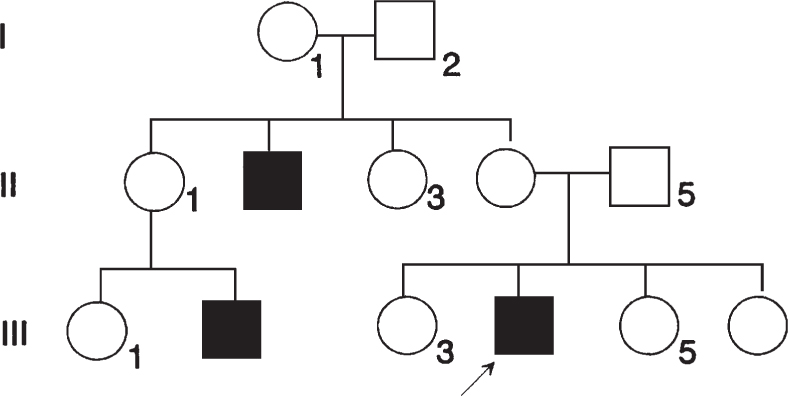
a. 100%
b. 75%
c. 50%
d. 25%
e. Virtually 0
261. Females occasionally have symptoms of X-linked recessive diseases such as Duchenne muscular dystrophy, hemophilia, or color blindness. Which of the following is the most common explanation?
a. Nonrandom lyonization
b. X-chromosome trisomy (47,XXX)
c. X autosome–balanced translocation that disrupts the particular X-chromosome locus
d. Turner syndrome (45,X)
e. 46,XY karyotype in a female
262. A Caucasian couple presents for genetic counseling after their first child, a son, is born with short limb dwarfism and diagnosed with autosomal dominant achondroplasia (MIM*100800). The physician obtains the following family history: the husband is the firstborn of four male children, and his next oldest brother has cystic fibrosis (MIM*219700). The wife is an only child, but she had DNA screening because a second cousin had cystic fibrosis and she knows that she is a carrier. There are no other medical problems in the couple or their families. The physician should now draw the pedigree with the female member of any couple on the left. The generations are numbered with Roman numerals and individuals with Arabic numerals; individuals affected with achondroplasia or cystic fibrosis are indicated. Which of the following risk figures applies to the next child born to this husband and wife?
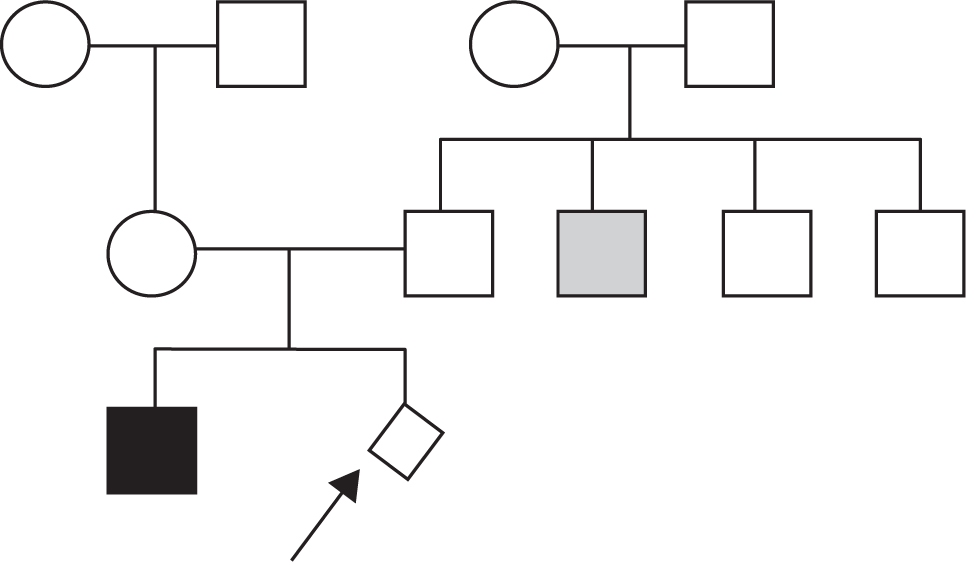
a. Achondroplasia ½, cystic fibrosis ¼
b. Achondroplasia ½, cystic fibrosis 1/8
c. Achondroplasia virtually 0, cystic fibrosis ¼
d. Achondroplasia virtually 0, cystic fibrosis 1/6
e. Achondroplasia virtually 0, cystic fibrosis 1/8
263. Isolated cleft lip and palate (meaning no other congenital anomalies are present) is a multifactorial trait. The recurrence risk of isolated cleft lip and palate is which of the following?
a. The same in all families
b. Not dependent on the number of affected family members
c. The same in all ethnic groups
d. The same in males and females
e. Affected by the severity of the cleft
264. Availability of DNA testing for many single disease traits has allowed routine prenatal screening of couples for disorders prevalent in their ethnic group. Which of the following genetic disorders has a similar incidence in different ethnic groups and would not be subject to different criteria for screening?
a. Cystic fibrosis
b. Thalassemias
c. Tay-Sachs disease
d. Down syndrome
e. Sickle cell anemia
265. In family with Charcot-Marie-Tooth disease (CMT), restriction analysis using sites flanking the CMT gene on chromosome 17 yields one large abnormal fragment and one smaller fragment that is seen in controls. What is the probable inheritance mechanism in this family?
a. X-linked recessive
b. Autosomal dominant
c. Autosomal recessive
d. Multifactorial
e. X-linked dominant
266. Many family studies employing DNA have the potential to demonstrate nonpaternity. If the physician ordering these analyses does not discuss this possibility with the couples involved, he or she is in violation of which of the following?
a. Patient confidentiality
b. Patient rights
c. Informed consent
d. Standards of care
e. Malpractice guidelines
267. The genesis of Prader-Willi syndrome by inheritance of two normal chromosomes from a single parent is a consequence of which of the following?
a. Germinal mosaicism due to paternal mutation
b. Genomic imprinting due to uniparental disomy
c. Chromosome deletion
d. Chromosome rearrangement
e. Anticipation
268. A child is born with spina bifida, a defect in the lower spinal cord and meninges that may cause bladder and lower limb dysfunction. The family history reveals that the father had a small spina bifida that was repaired by surgery. Which of the following is the most critical aspect of the medical evaluation as it pertains to genetic counseling?
a. A search for additional anomalies to determine if the child has a syndrome
b. A karyotype on the child
c. A serum folic acid level on the child
d. A spinal x-ray on the mother
e. A spinal x-ray on the father
269. A 1-year-old child develops fever and vomiting, and is unable to keep food down for 2 days. The physical examination discloses no congenital anomalies, and the baby resembles his parents. Which of the following laboratory findings are most likely if the child has a disorder of fatty acid oxidation?
a. Hypoglycemia, acidosis, and elevated urine dicarboxylic acids
b. Alkalosis and elevated serum ammonia
c. Acidosis and elevated urine reducing substances
d. Hypoglycemia, acidosis, and elevated serum leucine, isoleucine, and valine
e. Hepatomegaly, elevated serum liver enzymes, and elevated tyrosine
270. Laboratory tests on a sick child reveal a low white blood cell count, metabolic acidosis, increased anion gap, and mild hyperammonemia. Measurement of plasma amino acids reveals elevated levels of glycine, and measurement of urinary organic acids reveals increased amounts of propionic acid and methyl citrate. Which of the following processes is most likely?
a. Diabetes mellitus
b. A fatty acid oxidation disorder
c. Vitamin B12 deficiency
d. Propionic acidemia
e. A disorder in glycine catabolism
271. Neonatal screening is mandated in all states, but examines different numbers of diseases. Most commonly tested are phenylketonuria (PKU-MIM*261600), galactosemia (MIM*230400), congenital hypothyroidism, and sickle cell anemia (MIM*603903). Recently, a supplemental newborn screen using tandem mass spectrometry is being adopted by many states, allowing recognition of the more common organic acidemias and fatty acid oxidation disorders. Which of the following is the most important characteristic to qualify a disorder for newborn screening?
a. A highly accurate diagnostic test
b. A high frequency of disease
c. An advantage for treatment from early diagnosis
d. Use of microbial technology like the Guthrie method
e. A minimal incidence of false positive tests
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


