(1)
Toyonaka, Japan
1.1 From the First Report of Alzheimer’s Disease to Its Establishment as a Concept
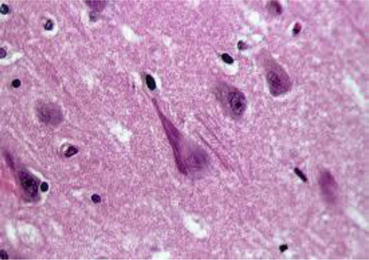
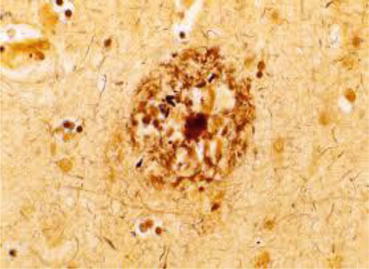
An illness, later to be named Alzheimer’s disease (AD), was first reported by the German psychiatrist Alois Alzheimer about 100 years ago (Fig. 1.1). On November 25, 1901, a 51-year-old woman named Auguste Deter (Fig. 1.2) was admitted to a municipal mental institution in Frankfurt am Main, where Alzheimer served as the department head. The letter of referral said, “The patient has suffered loss of memory, paranoia, insomnia, and a feeling of anxiety for a considerably long time. The patient is believed to be unable to deal with any kind of physical or mental labor.” It all began after Mrs. Deter strongly suspected her husband of having an extramarital affair. Her memory rapidly deteriorated thereafter. When she had to carry things, she could not remember where to carry them; therefore, she wandered back and forth in her apartment. In the end, she could not remember where to put things away, so she hid them in other places. She also had the delusion that someone was trying to kill her, so she would scream for hours. Alzheimer, who was exploring the anatomical basis of mental illnesses, felt that a “specific disease” was concealed behind the woman’s amnesia and pathological jealousy. He therefore examined her almost every day. However, she continued to become increasingly refractory to treatment; therefore, further treatment and examination became impossible. Alzheimer’s writings in the medical record ended in June 1902 with “When I try to examine Auguste Deter, she refuses, as before. She cries, shouts, and even hits me. She abruptly begins crying and often continues for several hours. So I sometimes must forcibly hold her down on the bed. She can no longer eat the meals provided to her. She has developed furuncles on her back.” Several months later, Alzheimer left the mental institution in Frankfurt. In April 1906, he received a phone call from the Munich Royal Psychiatric Hospital and learned that Auguste had died. He asked that her medical records and brain be sent to him. After suffering the disease for 4.5 years, she became bedridden and incontinent during the final days of her life. Auguste Deter died at the age of 55. Upon dissecting her brain, Alzheimer confirmed that it had atrophy, and numerous nerve cells in the cerebral cortex had disappeared. He discovered extensive neurofibrillary changes (Fig. 1.3) and senile plaques (Fig. 1.4). He concluded that these two changes comprised brain changes specific to AD. On November 3, 1906, at a meeting of the South-West German Psychiatrists held in Tübingen in southern Germany, Alzheimer presented Auguste’s case. This was the world’s first report on AD (Okamoto 2014).


Fig. 1.1
Alois Alzheimer
Fig. 1.2
Auguste Deter
In Europe at the end of the nineteenth century, dementia referred to either senile dementia or progressive paralysis. If a patient was aged, the disease was believed to be senile dementia; at middle age, the disease was considered progressive paralysis. In other words, a disease occurring in patients aged over 50 years was classified as senile dementia, a form of intellectual impairment that occurs with age. Auguste, the first known case of AD, was first examined at the age of 51. However, she was believed to have developed symptoms of disease in her 40s; thus, she would have ordinarily been diagnosed with progressive paralysis or a mental illness. Alzheimer, who was well versed in both progressive paralysis and mental illnesses, examined her symptoms, including memory disturbances, speech disruption, and a variety of other symptoms. He studied her brain after death and detected brain atrophy, senile plaques, and neurofibrillary changes. He concluded that she had a disease different from progressive paralysis or mental illness (Okamoto 2013). Emil Kraepelin named this illness “Alzheimer’s disease” and described it as a presenile dementia. He stated, “The pathological findings of the brain suggest it to be a type of severe senile dementia. In view of the fact that the patient had developed the disease in her 40s’, however, her disease cannot be said to be the same as senile dementia.” Kraepelin believed that because senile dementia occurred because of aging, there was no boundary between dementia and normal aging of the brain. The fact that the onset was early in life led him to conclude this was a special type of dementia.
It was later revealed that the brain of patients with senile dementia had senile plaques, but the degree of neurofibrillary changes and cerebral atrophy tended to be lesser. The disease progressed slowly, as did the symptoms; therefore, AD was believed to be distinct from senile dementia. However, some declared that based on clinical and pathological data, AD and senile dementia were the same clinical entity with age of onset being the only difference. Others stressed that since dementia occurs with aging, it should be categorized as a “condition,” not a disease. During the 1970s, after lengthy debates, scientists concluded that AD and senile dementia should not be considered separately but be diagnosed together as AD (Okamoto 2014).
1.2 Elucidating the Formation of Neurofibrillary Changes
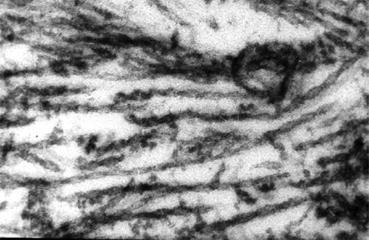
Michael Kidd discovered paired helical filaments (PHFs) (Fig. 1.5), comprised of two twisted filaments, as the major structural component of neurofibrillary changes or tangles (Kidd 1963). To elucidate neurofibrillary changes, it was necessary to identify the structural components of PHFs. Robert Terry et al. succeeded in doing this using a biochemical strategy (Terry et al. 1964). Khalid Iqbal and Yasuo Ihara then found that this structural component was the tau protein, one of microtubule associated proteins, with abnormal phosphorylation (Grundke-Iqbal et al. 1986; Ihara et al. 1983). Inside the nerve cells, there are proteins in the form of microtubules acting as the cytoskeletal structure. Tau stabilizes microtubules by attaching and detaching itself from them by phosphorylation level. Abnormal phosphorylation causes tau protein to separate from the microtubules. Free tau protein aggregates to form a β-sheet structure that forms PHFs, leading to neurofibrillary changes. Researchers believe that this in turn destroys microtubules and nerve cells, triggering AD.
1.3 The Acetylcholine Hypothesis
It is well known that Parkinson’s disease occurs because of a reduction in levels of the neurotransmitter dopamine. Several research groups suspected that the secretion of neurotransmitters might also be involved in causing AD; therefore, they investigated the secretion of acetylcholine, a neurotransmitter involved with memory. They found that acetylcholine secretion decreased drastically in the AD brain and observed the following: (1) reduced choline acetyltransferase activity (Bartus et al. 1982), (2) a deficit of cholinergic nerves in the basal forebrain (Whitehouse et al. 1982), and (3) a reduction in nicotinic acetylcholine receptors (Sihver et al. 1999). This led to the establishment of the “acetylcholine hypothesis,” which claims a reduction in acetylcholine is the cause of AD. If a person develops AD, neurofibrillary tangles occur at the early stages in brain regions originally containing large amounts of acetylcholine. This fact appeared to confirm the hypothesis. Supporters of this hypothesis believed that if a reduction in acetylcholine caused AD, then replenishing acetylcholine might help. They therefore attempted a method of treatment that involved supplementing with substances that are metabolic precursors of acetylcholine. They believed that since providing l-DOPA, which is converted to dopamine inside the brain, to treat Parkinson’s disease was proving successful, the same principle might also apply to AD. Unfortunately, the treatment was ineffective because other neurotransmitters, not just acetylcholine, were found to be decreased in the AD brain (Okamoto 2014). As a result, the acetylcholine hypothesis, which attracted enthusiastic support in the latter half of the 1970s, exited from mainstream research. Nevertheless, activation of acetylcholine is still considered an important strategy in the treatment of AD. A leading example is the use of inhibitors of acetylcholinesterase, such as tacrine, donepezil, galantamine, and rivastigmine.
1.4 The Amyloid Hypothesis
During the 1980s, AD researchers made significant progress investigating senile plaque. Senile plaques are deposits of amyloid outside nerve cells. They are also deposited in the cerebral blood vessels of patients with AD and Down syndrome. George G. Glenner at the University of California took note of this fact. He isolated amyloid from the cerebral vascular walls of subjects with AD and those with Down syndrome, purified it, and determined its amino acid sequence. In 1984, Glenner and Wong named the peptide they discovered β-amyloid (Glenner and Wong 1984). In 1985, Konrad Beyreuther et al. in Germany also revealed β-amyloid in the nuclei of senile plaques to be the same as that which Glenner had discovered (Masters et al. 1985). β-Amyloid was found to be comprised of over 40 amino acids linked together, and senile plaques were a hardened aggregation of β-amyloid. β-Amyloid was suspected to be a fragment of an even larger protein. In 1987, a group led by Jie Kang et al. in Germany identified amyloid precursor protein (APP). APP was found to be present inside brain cells, and β-amyloid was a fragment of APP cleaved by enzymes. They also found that the APP was a transmembrane protein. Moreover it was discovered that the APP gene was on chromosome 21 (Kang et al. 1987). This identification of APP resulted in the emergence of the amyloid hypothesis, which states that AD begins when there is an abnormal increase in β-amyloid (Okamoto 2014).
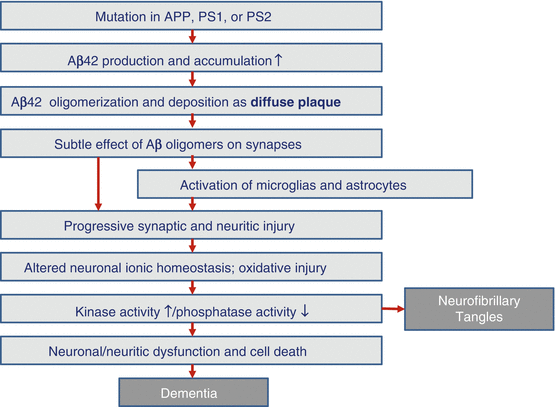
APP comprises about 700 amino acids. It was found that the phenomenon which causes APP fragmentation to create β-amyloid occurs very slowly even in the normal human brain (Haass et al. 1992). However, due to genetic abnormalities or other reasons, β-amyloid may end up being produced in large amounts. This led to the emergence of the “amyloid hypothesis” which states that senile plaques result from accumulation of β-amyloid in bulk outside nerve cells; this damages nerve cells and triggers neurofibrillary tangles that in turn cause neuropathy, nerve cell death, and neurologic deficits. Dementia is hypothesized to develop as a result of this series of events (Hardy and Selkoe 2002) (Fig. 1.6). However, the role of APP in human metabolism as well as the normal action of β-amyloid was unknown (Okamoto 2014).
The amyloid hypothesis has predominated AD research due to a major discovery that helped make this hypothesis rock-solid: the discovery by John Hardy in the UK of a mutation in the APP gene. Although most AD cases are not familial, approximately 10 % are of familial onset (Okamoto 2014). Many researchers have therefore attempted to identify the causal gene of familial AD and based on this research have tried to investigate the cause of general AD that develops in old age (i.e., “sporadic AD”). Hardy studied the brains of patients with familial juvenile AD and in 1991 discovered a characteristic APP gene mutation (Goate et al. 1991; Chartier-Harlin et al. 1991). Most types of β-amyloid that deposit on senile plaques have either 40 (Aβ40) or 42 amino acids (Aβ42). The type comprising 42 amino acids is especially prone to agglutination. It was shown that if the APP gene had mutations of the type Hardy discovered, then β-amyloid with 42 amino acid sequences becomes even more prone to being cleaved, making it easier for β-amyloid to deposit and form senile plaques at an early stage. Consequently nerve cells are damaged and develop neurofibrillary tangles under these conditions. The fact that a person subject to this cascade develops dementia while still young provided good evidence to support the amyloid hypothesis (Hardy and Higgins 1992).
However, even among families with familial AD, extremely few individuals had the APP gene mutation (Okamoto 2014). Researchers continued to expand their studies, strongly suspecting the presence of other causal genes. In 1995, a gene named presenilin 1 was discovered by Peter St. George-Hyslop in Canada (Sherrington et al. 1995). This was followed by the discovery of another gene which he named presenilin 2 (Sherrington et al. 1996).
A cleavage enzyme is required at two locations to cut β-amyloid from APP. The enzyme at the first location is called β-secretase and at the second location is γ-secretase (Fig. 1.7). The two presenilins St. George-Hyslop had discovered were found to work as γ-secretases. If presenilin has a genetic mutation, it becomes liable to cleave Aβ 42. The cleavage of Aβ 42 continues to be stepped up: it accumulates, forms senile plaques, and eventually causes dementia. This process is similar to the APP genetic mutation. Therefore, mutations in the APP and two presenilin genes cause steady accumulation of β-amyloid and generate the pathology of familial AD. It was also supposed that even sporadic AD was also caused by β-amyloid accumulation as the patoent ages (Okamoto 2014). As described, the discovery of presenilin led to understanding that dementia originates with β-amyloid, further strengthening the amyloid hypothesis.
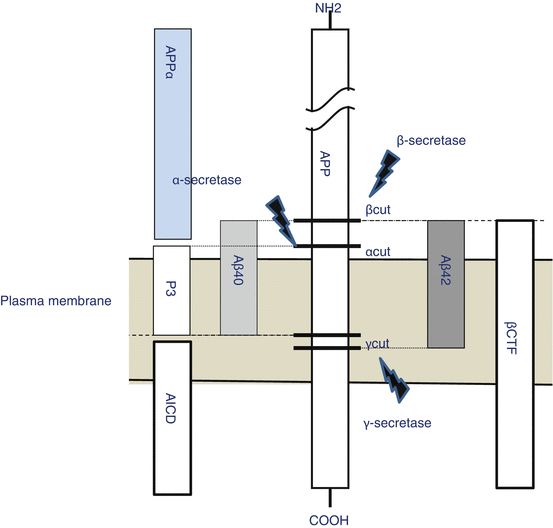
Fig. 1.7
APP processing. β-Cut by β-secretase and γ-cut by γ-secretase generate Aβ40 or Aβ42. In physiological condition, α-cut by α-secretase predominantly causes extracellular release of APPα. It is supposed that combinations of these cuttings generate P3, AICD, or βCTF
On the other hand, α-secretase which cleaves within the fragment of β-amyloid was also identified (Sisodia 1992). The α-secretase pathway is the predominant APP processing pathway. Thus, α-secretase cleavage precludes β-amyloid formation and is considered to be the non-amyloidogenic pathway of APP processing in the physiological condition (Fig. 1.7). α-Secretase is a member of the ADAM (“a disintegrin and metalloprotease domain”) family, which are expressed on the surfaces of cells. α-Secretases cleave APP to release its extracellular domain – a fragment known as APPs – into the extracellular environment (Lammich et al. 1999).
1.5 Development of Disease-Modifying Drugs (DMDs) Based on the Amyloid Hypothesis
1.5.1 Amyloid Vaccination
According to the amyloid hypothesis, one only needs to remove the amyloid that has deposited in the AD brain. The unique idea of removing amyloid by vaccination began in 1999 after Schenk et al. conducted an experiment in which they administered synthesized amyloid peptide (Aβ42) and adjuvant to AD model mice. With this amyloid vaccination, senile plaques were shown to decrease in model mice, and the formation of new senile plaques was prevented (Schenk et al. 1999). The following year, a clinical study on active immunity using an Aβ42 vaccine began. Unfortunately, the test was suspended when 6 % of the subjects suffered cerebral meningitis. One patient was diagnosed with T-cell cerebrospinal meningitis. Cerebral meningitis caused by the active immunity vaccine was believed to have an autoimmune cause, triggered by T-cells reacting to amyloid. Therefore, researchers came to consider administering a vaccine using antigens without the Aβ domain that activate T-cells, such as a passive injectable vaccine comprised of exogenously manufactured human anti-amyloid antibodies (Monsonego et al. 2003).
Regarding the effects of active immunity vaccines, it was discovered from autopsies of subjects who had died that although the volume of senile plaques decreased, progression of dementia could not have been suppressed. A long-term follow-up survey of the study subjects showed no differences from the placebo group in terms of survival rate or rate of dementia progression (Holmes et al. 2008).
At present, the focus of development has shifted to passive immunity vaccine therapy. Because anti-amyloid antibodies are administered intravenously, they react directly with amyloid deposited on the cerebrovascular wall, causing adverse events such as vascular edema. This has become a problem. The fact that the body readily produces antibodies against the monoclonal antibodies administered, and that excessive costs are incurred due to the need for repeated administration to maintain the effect of the antibodies, has also become a problem. Despite these drawbacks, massive development costs have been invested in developing amyloid vaccines; however, none of the clinical trials has come to a successful conclusion.
1.5.2 γ-/β-Secretase Inhibitors
1.5.2.1 γ-Secretase
Gamma (γ)-secretase is a membrane protein complex comprised of presenilin (PS), nicastrin, APH-1, and Pen 2. γ-Secretase is an aspartic protease with low substrate specificity. However, when cleaving Aβ from APP-βCTF, which has just β-cut, cleavage occurs predominantly at the 40th residue from the N-terminal. Ordinarily, Aβ40 accounts for 80–90 % of the total amount of Aβ produced, and Aβ42 accounts for 10–20 % (Fig. 1.7). Recent research shows Aβ40 and Aβ42 are cleaved in stages, each via separate pathways. More specifically, ε-cleavage first occurs by γ-secretase six to nine residues closer to the C-terminal on the Aβ40 or Aβ42 side than has been conventionally reported, and δ- and γ-cleavage appears to consequently occur thereafter. As shown in the Figure 1.8, Aβ is produced via two pathways, namely, the Aβ49 → 46 → 43 → 40 → 37 route and the Aβ48 → 45 → 42 → 38 route, with the latter anticipated to be more pathological (Kakuda et al. 2012) (Fig. 1.8). Aβ38 was also revealed to be produced not only from Aβ42 but also from Aβ43. These two routes are believed to cross each other (Okochi et al. 2013).
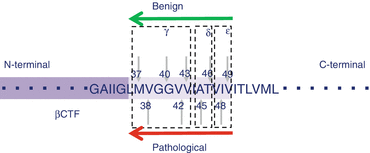
Fig. 1.8
Cleavage by γ-secretase. γ-secretase has multiple cleave sites of βCTF, i.e., ε-cleavage, δ-cleavage, and γ-cleavage. There are two pathways, i.e., the Aβ49 → 46 → 43 → 40 → 37 route and the Aβ48 → 45 → 42 → 38 route. The former is considered to be a benign pathway, and the latter, a pathological pathway
Past reports have shown there are over 90 γ-secretase substrates (Haapasalo and Kovacs 2011). Of these, Notch 1 is a substrate as important as APP. It has been established that a Notch 1 phenotype is observed in PS knockout mice (in which γ-secretase has been eliminated); it causes severe abnormalities (Shen et al. 1997). Notch signaling plays an important role in cell interactions during brain development (Artavanis-Tsakonas et al. 1999). Notch is also known to work as a proto-oncogene, tumor-suppressing molecule, in certain types of cancer (Lobry et al. 2011). In addition, Notch signaling reportedly plays an important role in the maintenance and differentiation of nerve stem cells, the structure of nerve tissues, and synaptic plasticity (Louvi and Artavanis-Tsakonas 2006; Ables et al. 2011). Therefore, impeding the physiological roles of Notch signaling by γ-secretase inhibitors (GSIs) could have serious side effects.
1.5.2.2 BACE1 (Beta-Site APP Cleaving Enzyme 1)
β-Secretase was identified as BACE1, and a knockout mouse was produced. Since no notable phenotypes were recognized in this knockout mouse, hopes arose that BACE1 inhibitors could be used as DMDs to treat AD with a minimum of side effects (Luo et al. 2001). It gradually became apparent, however, that BACE1 was required for peripheral nerve myelination by Schwann cells and correct formation of nerve axons, thus indicating a risk of side effects (Willem et al. 2006). Unlike other aspartic proteases, BACE1 has a large active site and few hydrophobic residues. Researchers therefore point out that the molecular weight of inhibitor drugs would be high, making it difficult for them to pass through the blood–brain barrier.
1.5.2.3 Current Status and Problems of GSI Development
On August 17, 2010, Eli Lilly and Company announced they had discontinued the development of semagacestat, a nonselective GSI. This was because the preliminary-stage results of two long-term phase III clinical tests for this drug showed no suppressive effect on the advancement of mild to moderate-degree AD. In addition, the drug was accompanied by aggravation of cognitive function scores (ADAS-cog), reduced patients’ abilities in daily living, and promoted skin cancer. The news was received with a huge shock, because the clinical trials involved over 3,000 patients worldwide and had been carried out with high hopes and at a huge expense. Since then, although clinical trials have been conducted on drugs with a similar mechanism of action as semagacestat, no successful evidence has been obtained as yet.
Besides the blocking of Notch signaling, nonselective GSIs are anticipated to reduce production of Aβ40, Aβ42, and the intracellular domain of APP (AICD). On the other hand, they are also anticipated to cause an increase in βCTF (the C-terminal fragment of APP that has been cleaved by β-secretase) (Fig. 1.7). Research suggests Aβ40 effectively prevents the aggregation of Aβ42 (Kim et al. 2007). Therefore, reducing Aβ40 using nonselective GSIs carries the risk of promoting amyloid deposition rather than inhibiting it. One report claims that the total volume of Aβ, rather than the Aβ42/Aβ40 ratio, defines the age of onset of familial Alzheimer’s disease (FAD) (Kumar-Singh et al. 2006). There is a possibility that nonselective GSIs may reduce the total volume of Aβ, which might actually promote the pathology of AD. AICD corresponds to the intracellular domain of Notch receptor and is believed to physiologically function in controlling nerves and synapses (Zheng and Koo 2011). It can be readily assumed, therefore, that if AICD decreases because of nonselective GSI, it would lead to some form of cognitive dysfunction. Nonselective GSIs also have the potential to increase βCTF levels. Some researchers have reported that βCTF has neurotoxicity (Yankner et al. 1989); therefore, its increase may also lead to neurological disorders.
1.5.2.4 Expectations from γ-Secretase Modulators (GSMs)
It has been reported from epidemiological research into AD that certain nonsteroidal anti-inflammatory drugs (NSAIDs) specifically impede Aβ42 production, but do not block the production of Aβ40 or Notch signaling. This Aβ42-suppression effect was also shown to be independent of the cyclooxygenase (COX) inhibitory activity of NSAIDs (Weggen et al. 2001). Because these compounds “adjust” γ-secretase activity specifically at the γ-cleavage sites, they are called GSMs and are increasingly of interest as treatment drugs that do not create the adverse reactions caused by Notch inhibition.
GSMs reportedly show the following characteristics: (1) they suppress production of Aβ42, (2) promote the production of short Aβ (Aβ38 and Aβ37), (3) do not change the total Aβ or βCTF production amount, and (4) do not affect Notch signaling (Crump et al. 2013). Based on this definition, NSAIDs such as ibuprofen, indomethacin, sulindac sulfide, flurbiprofen, and their analogs (first-generation GSM) have been tested in clinical trials. Because of their weak Aβ42-inhibitory effects and poor transferability to the brain, clinical trials did not proceed well. More recently, development has been under way on second-generation GSMs featuring improved Aβ42-inhibitory effects and better transferability to the brain.
The debate continues about the mechanism by which the effects of GSMs are manifested. Within the Aβ-production route beginning with ε-cleavage, GSMs delay the separation between Aβ42 and γ-secretase and promote cleavage to Aβ38. In contrast, PS1 mutation or GSMs with reverse actions (elevating Aβ42 in an opposite manner) can shorten the separation between this enzyme and its substrate (Okochi et al. 2013). This finding shows that simply inhibiting γ-secretase brings about a reverse effect on the treatment of AD, raising hopes for the future development of GSMs.
1.5.2.5 Reexamination of the Amyloid Cascade Hypothesis
The stagnation of clinical trials of GSIs and other DMDs questions the practicality of the amyloid cascade hypothesis as a drug discovery strategy. Even if the amyloid cascade hypothesis is correct, recent findings suggest that it takes about 20 years from the start of the appearance of amyloid and its deposition to the manifestation of dementia (Jack et al. 2010). Therefore, even if the AD is mild, amyloid has been deposited over a wide area. Therefore, it is easy to assume that even if DMDs had been administered during this period, their effects would be extremely limited. In contrast, a concept called preclinical AD is increasingly being advocated. In other words, even though symptoms of dementia may not have manifested, if amyloid abnormalities are revealed by amyloid imaging and are present in the cerebrospinal fluid, physicians are increasingly likely to identify the disease as AD and initiate treatment by aggressively prescribing DMDs (Mangialasche et al. 2010).
According to the amyloid cascade hypothesis, if the rise in Aβ levels and its deposition could be suppressed, it would be possible to treat AD. Is this really true? It has recently been shown that tau is needed for Aβ to cause neuropathy. Transgenic mice expressing mutant APP show memory and learning disabilities in a water maze. If these mice are crossed with tau knockout mice, the tau gene-defective offspring have no memory or learning disabilities. They show no differences from controls, even though amyloid has been deposited (Roberson et al. 2007). On the other hand, if fibrillized Aβ42 is injected into the brains of transgenic mice expressing frontotemporal lobar dementia FTDP-17 mutant (P301L) tau, the fibrous accumulation of tau is stepped up (Gotz et al. 2001). These animal experiments suggest Aβ associates with tau protein to advance the pathological process. Suppressing the pathology of Aβ only, using DMDs, therefore may be only part of the story.
In pathological research on AD, it has primarily been thought that the number of neurofibrillary changes (tau pathology) is related to the duration of the disease and clinical symptoms (Braak and Braak 1996). After 1998, variations in diverse tau genes were successively reported in families with dementia that essentially had tauopathies only (Lee et al. 2005). These diseases are generally referred to as frontotemporal dementia with Parkinsonism linked to chromosome 17 (FTDP-17), revealing the existence of a mechanism by which tauopathy is clearly different from the amyloid cascade hypothesis. Thus, it is possible the development of tauopathy is more closely linked to nerve cell death than to β-amyloid, regardless of the accuracy of the amyloid cascade hypothesis that claims the formation of tauopathy takes place downstream of β-amyloid accumulation.
1.6 Paradigm Shift of the Dementia Pathology Hypothesis
1.6.1 The Pathology of Abnormal Protein Accumulation Commonly Seen in Dementia
Dementia pathologies caused by neurodegeneration are commonly explained by the mechanism of abnormal protein accumulation inside nerve cells. Until now, it has been possible to cite pathologies such as amyloidopathy, tauopathy, TDP-43 proteinopathy, α-synucleinopathy, polyglutamine disease, and prion disease. In all these diseases, it is predicted that abnormal protein is subject to phosphorylation and ubiquitination, and fibers aggregate and acquire nerve cell toxicity.
1.6.1.1 Amyloidopathy (For Details, See the Previous Section)
AD’s pathological process can be explained by amyloidopathy. Aβ is a highly cohesive protein with a molecular weight of approximately 4,000 kDa; it forms senile plaques and cerebrovascular amyloids. APP is a type I transmembrane protein and is cleaved by α-secretase, β-secretase, and γ-secretase. However, Aβ is formed by phased cleavage of β-secretase and γ-secretase (Fig. 1.7). Two primary molecular species of Aβ are produced: Aβ40, which has 40 amino acid residues, and the highly cohesive Aβ42 that is two residues longer. Aβ42 deposits at an early stage and forms senile plaques, the characteristic pathological change seen in AD.
Aβ is secreted extracellularly and broken down by neprilysin (Iwata et al. 2000) and insulin-degrading enzyme. The part of the secreted Aβ then begins to form amyloid fibrils and aggregates. It either deposits in the intercellular space of the cerebral parenchyma to form senile plaques or deposits on the cerebrovascular wall and forms amyloid angiopathy. In the past, the Aβ of these insoluble aggregates was believed to be neurotoxic. More recently, however, Aβ oligomer present in soluble form, before fibrous aggregation takes place, is believed to demonstrate even stronger toxicity.
1.6.1.2 Tauopathy
The pathological process of tauopathy consists of a disease condition characterized by abnormal accumulation of tau on nerve and glial cells. It is not only seen in AD neurofibrillary changes but also in degenerative diseases such as Pick’s disease, corticobasal degeneration, and progressive supranuclear palsy.
Tau, a microtubule-associated protein, promotes the formation of microtubules. It is expressed as six isoforms by selective splicing. It can be composed of three (R1, R3, and R4) or four repeat regions (R1, R2, R3, and R4), those that have both exon 2 (E2) and 3 (E3), those that have E2 only, and those that have neither E2 nor E3 (Fig. 1.9). Tau bindings to microtubules are formed by these repeat regions.
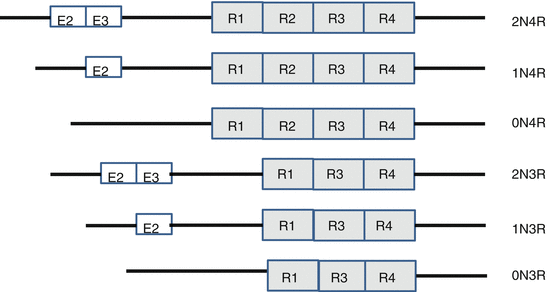
Fig. 1.9
Isoforms of tau. Six isoforms of tau are composed of three (R1, R3, and R4) or four repeat regions (R1, R2, R3, and R4), those that have exon 2 (E2) and 3 (E3), those that have exon 2 only, and those that have neither exon 2 nor 3
Tau contains numerous potential phosphorylation sites. In solution, it does not adopt a folded structure, making it an ideal target for intracellular kinases. In fact, the polymerization and depolymerization processes seen with microtubules are caused by the phosphorylation and dephosphorylation reactions. Tau that has been excessively phosphorylated loses its microtubule-binding capabilities, making the microtubules unstable. Tau that has separated from its associated microtubules rises to high intracytoplasmic concentrations, undergoes self-polymerization, and causes neurofibrillary changes. PHFs and straight filaments, which are constituent factors of AD neurofibrillary changes, are fibrils made of excessively phosphorylated tau. Kinases related to tau phosphorylation include cyclin-dependent kinase 5 (Cdk5), glycogen synthase kinase 3 (GSK3), mitogen-activated protein kinase (MAPK), and stress-activated protein kinase (SAPK).
Accumulation of tau protein is also observed in Pick’s disease, corticobasal degeneration, and progressive supranuclear palsy; therefore, these pathologies have come to be regarded together as tauopathies. There are three types of tauopathy: (1) 3-repeat type (in which insoluble 3-repeat tau predominates, such as in Pick’s disease and FTDP-17), (2) 4-repeat type (insoluble 4-repeat tau predominates, such as corticobasal degeneration and progressive supranuclear palsy), and (3) 3 + 4 repeat type (mixed type such as AD and neurofibrillary change-type dementia). Many aspects of the disease specificity of tau isoforms remain unclear. However, isoforms require a set physiological ratio, and the disruption of balances may lead to disease onset. As mentioned previously, development of a therapeutic method based on the amyloid cascade hypothesis is currently facing difficulties, suggesting the need to develop treatment drugs based on tauopathy as well.
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


