Basic Principles
Basic principles are often perceived as the most challenging aspect of learning pharmacology. While possibly conceptually difficult, this subject is absolutely key to gain understanding how medications exert their effects and side effects. While licensing exams may not stress basic principles in their pure form, this topic is at the core of many pharmacology questions that may appear in these examinations. Therefore, a strong knowledge of basic principles will help you with your study of pharmacology, and throughout your medical career.
PHARMACOKINETICS
Define the following terms:
Pharmacokinetics
Field of study that deals with time required for drug absorption, distribution in the body, metabolism, and method of excretion. In short, it is the body’s effect on the drug.
Volume of distribution(Vd)
The apparent volume in the body available to contain the drug. Formula: Vd = Dose/Plasma Drug Concentration
Is Vd a physiologic value?
No
Is Vdan absolute value for any given drug?
No
Clearance (CI)
Volume of blood cleared of the drug per unit time; Cl = Rate of elimination of drug/plasma drug concentration; Total Body Cl = Clhepatic + Clrenal + Clpulmonary + Clother
Half-life (t1/2)
Time required for plasma concentration of drug to decrease by one-half after absorption and distribution are complete; t1/2 = (0.693 × Vd)/(Cl)
Bioavailability (F)
The fraction of (active) drug that reaches the systemic circulation/site of action after administration by any route; F = (AUCpo)/(AUCiv), where AUCpo and AUCiv are the extravascular and intravenous areas under the plasma concentration versus time curves, respectively
Steady state (Css)
Steady state is reached when the rate of drug influx into the body = the rate of drug elimination out of the body; Css = plasma concentration of drug at steady state
How much drug is left after two half-lives?
25%
How much drug is left after three half-lives?
12.5%
During constant infusion, what percent of steady state is reached after one half-life?
50%
During constant infusion, what percent of steady state is reached after two half-lives?
75%
During constant infusion, what percent of steady state is reached after three half-lives?
87.5%
During constant infusion, what percent of steady state is reached after four half-lives?
94%
Give the equation for the following terms:
Infusion rate (k0)
k0= Cl × Css
Loading dose (LD)
LD = (Vd × CSS)/(F); for examination purposes, F is usually 1
Maintenance dose (MD)
(Cl × Css × τ)/(F), where τ (tao) is the dosing interval
Clearance (Cl)
Cl = K × Vd, where K is the elimination constant
Volume of distribution (Vd)
Vd= = (LD)/(CSS)
Half-life (t1/2)
tl/2 = (0.693)/(K) or (0.693 × Vd)/(Cl)
What happens to the steady state concentration of a drug if the infusion rate is doubled?
Steady state concentration is also doubled; remember that dose and concentration are directly proportional (linear kinetics); Css × k0/Cl
If there is no active secretion or reabsorption, then renal clearance (Clrenal) is equal to what?
Glomerular filtration rate (GFR)
If a drug is protein bound, then Clrenal is equal to what?
GFR × free fraction (of drug)
What happens to the LD in patients with impaired renal or hepatic function?
Stays the same
What happens to the MD in patients with impaired renal or hepatic function?
Decreases
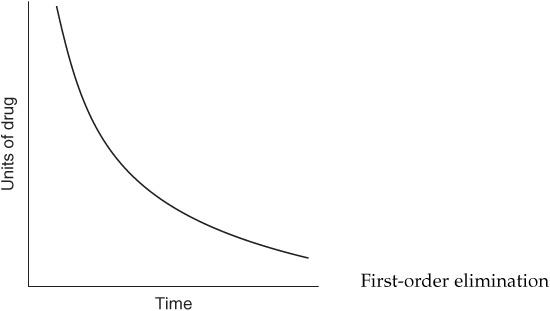
For each of the following statements, state whether it refers to zero-order elimination or first-order elimination?
Rate of elimination is constant, regardless of concentration
Zero-order elimination
Plasma concentration decreases exponentially with time
First-order elimination
Rate of elimination is proportional to the drug concentration
First-order elimination
Plasma concentration decreases linearly with time
Zero-order elimination
Rate of elimination is independent of concentration
Zero-order elimination
Rate of elimination is dependent on concentration
First-order elimination
What are some examples of drugs/substances that undergo zero-order elimination?
Acetylsalicylic acid (Aspirin, ASA) at high/toxic concentrations; phenytoin; ethanol
Describe the following types of metabolism:
Phase I metabolism
Metabolism that generally yields more polar, water-soluble metabolites (may still be active); enzyme activity decreases with patient’s age
Phase II metabolism
Metabolism that generally yields very polar, inactive metabolites (renally excreted); enzyme activity does not decrease with patient’s age
Phase I (microsomal) metabolism involves what types of reactions?
Oxidation; reduction; hydrolysis (carried out by cytochrome P-450 enzymes)
Phase II (nonmicrosomal) metabolism involves what types of reactions?
Glucuronidation; acetylation; sulfation; amidation; glutathione conjugation
Give examples of drugs that undergo phase II metabolism:
Isoniazid (INH), morphine, 6-mercaptopurine, acetaminophen
What are the potential consequences of phase I oxidation reactions with regard to drug activity and elimination?
Drug activity may or may not change (no rule, ie, potentially dangerous outcome). Drug elimination is usually increased due to greater water solubility.
What are the potential consequences of phase II reactions with regard to drug activity and elimination?
Drug products of phase II reactions are usually inactive and their renal elimination is enhanced.
Where are cytochrome P-450 enzymes found?
Smooth endoplasmic reticulum of cells in mainly the liver, but also found in the gastrointestinal (GI) tract, kidney, and lungs
Explain what each type of the following clinical phases in drug development is trying to accomplish?
Phase I
Safety in healthy individuals; drug pharmacokinetics
Phase II
Efficacy in diseased individuals (small scale trials, single- double-blind)
Phase III
Efficacy in diseased individuals (small scale trials, single- or double-blind)
Phase IV
Postmarketing surveillance (monitored release)
At what point during drug development is an investigational new drug (IND) application filed?
Before phase I
At what point during drug development is a new drug application (NDA) filed?
After phase III (and before phase IV)
What does the term bioequivalence mean?
When comparing two formulations of the same compound, they are said to be bioequivalent to each other if they have the same bioavailability and the same rate of absorption.
What is the first-pass effect?
After oral administration, many drugs are absorbed intact from the small intestine and transported first via the portal system to the liver, where they undergo extensive metabolism, therefore usually decreasing the bioavailability of certain oral medications.
How many liters are in each of the following compartments of an average adult human?
Blood
5L
Plasma
3L
Total body water (TBW)
42 L (avg. 70 kg man × 60%. For women 50% of mass [body weight] in kg is body water due to lower lean muscle mass and higher fat content [adipose tissue]).
What is the most common plasma protein that drugs bind to?
Albumin
Displacing a drug that is bound to plasma protein(s), for example, albumin, will increase its what?
Its free fraction (therefore may possibly increase the risk of toxicity because the plasma concentration of active drug has been increased, yet depending on the drug, an increase in free fraction may actually increase its metabolism because more drug is available to metabolizing enzymes)
For each of the following mechanisms of membrane transport, state if energy is required, if a carrier is required, and if the system is saturable?
Passive diffusion
No energy required; no carrier; not saturable (proportional to concentration gradient)
Facilitated diffusion
No energy required; carrier required; saturable
Active transport
Against concentration/electrical gradient, therefore energy required; carrier required; saturable
The permeation of drugs across cellular membranes is dependent on what drug properties and (local) circumstances?
Drug solubility; drug concentration gradient; drug ionization; surface area; vascularity
Acidification of urine will increase renal elimination of what types of drugs?
Weak bases (ionized form of drug, BH+) will be trapped in the renal tubules and thus excreted in the urine.
Acidification of urine will decrease renal elimination of what types of drugs?
Weak acids (nonionized, HA, form of a drug) can cross membranes.
Alkalinization of urine will increase renal elimination of what types of drugs?
Weak acids (ionized, A−, form of a drug) will be trapped in the renal tubules and thus excreted in the urine.
Alkalinization of urine will decrease renal elimination of what types of drugs?
Weak bases (nonionized, B, form of a drug) can cross membranes.
What agents are used to acidify urine?
NH4Cl; high dose of vitamin C
What agents are used to alkalinize urine?
NaHCO3; acetazolamide
Give an example of a weakly acidic drug:
Acetylsalicylic acid (ASA); barbiturates
Give an example of a weakly basic drug:
Amphetamines
Give examples of drugs metabolized by each of the following cytochrome P-450 enzymes metabolizes:
CYP 1A2
Caffeine; ciprofloxacin; theophylline; R-warfarin
CYP 2C9
Ibuprofen; naproxen; phenytoin; S-warfarin
CYP 2C19
Diazepam; omeprazole
CYP 2D6
Codeine; dextromethorphan; fluoxetine; haloperidol; loratadine; metoprolol; paroxetine; risperidone; thioridazine; venlafaxine
CYP 2E1
Ethanol; INH; acetaminophen (at high doses)
CYP 3A4 (50%-60% of all therapeutically used drugs are metabolized via CYP 3A4)
Alprazolam; carbamazepine; cyclosporine; diltiazem; erythromycin; fluconazole; itraconazole; ketoconazole; lidocaine; lovastatin; midazolam; nifedipine; quinidine; simvastatin; tacrolimus; verapamil
Give examples of drugs and herbal extracts that generally induce cytochrome P-450 enzymes:
Phenobarbital; nicotine; rifampin; phenytoin; carbamazepine; St. John’s wort; chronic ethanol consumption
Give examples of drugs and foods that generally inhibit cytochrome P-450 enzymes:
Erythromycin; ketoconazole; ciprofloxacin; quinidine; cimetidine; omeprazole; ritonavir; chloramphenicol; acute alcohol intoxication, grapefruit juice
The following graph depicts what type of elimination?
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


