Chapter 15 Assessing drug safety
Introduction
Safety is addressed at all stages in the life history of a drug, from the earliest stages of design, through pre-clinical investigations (discussed in this chapter) and preregistration clinical trials (Chapter 17), to the entire post-marketing history of the drug. The ultimate test comes only after the drug has been marketed and used in a clinical setting in many thousands of patients, during the period of Phase IV clinical trials (post-marketing surveillance). It is unfortunately not uncommon for drugs to be withdrawn for safety reasons after being in clinical use for some time (for example practolol, because of a rare but dangerous oculomucocutaneous reaction, troglitazone because of liver damage, cerivastatin because of skeletal muscle damage, terfenadine because of drug interactions, rofecoxib because of increased risk of heart attacks), reflecting the fact that safety assessment is fallible. It always will be fallible, because there are no bounds to what may emerge as harmful effects. Can we be sure that drug X will not cause kidney damage in a particular inbred tribe in a remote part of the world? The answer is, of course, ‘no’, any more than we could have been sure that various antipsychotic drugs – now withdrawn – would not cause sudden cardiac deaths through a hitherto unsuspected mechanism, hERG channel block (see later). What is not hypothesized cannot be tested. For this reason, the problem of safety assessment is fundamentally different from that of efficacy assessment, where we can define exactly what we are looking for.
Here we focus on non-clinical safety assessment – often called preclinical, even though much of the work is done in parallel with clinical development. We describe the various types of in vitro and in vivo tests that are used to predict adverse and toxic effects in humans, and which form an important part of the data submitted to the regulatory authorites when approval is sought (a) for the new compound to be administered to humans for the first time (IND approval in the USA; see Chapter 20), and (b) for permission to market the drug (NDA approval in the USA, MAA approval in Europe; see Chapter 20).
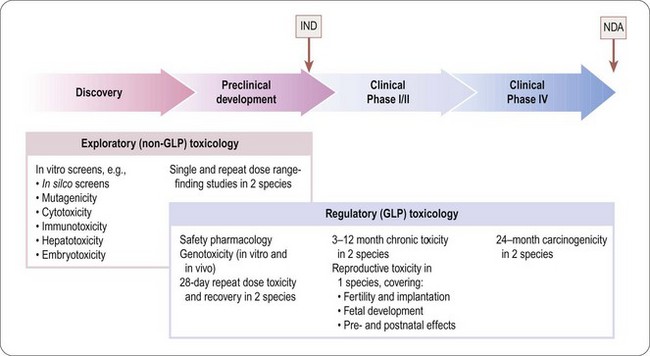
The programme of preclinical safety assessment for a new synthetic compound can be divided into the following main chronological phases, linked to the clinical trials programme (Figure 15.1):
• Exploratory toxicology, aimed at giving a rough quantitative estimate of the toxicity of the compound when given acutely or repeatedly over a short period (normally 2 weeks), and providing an indication of the main organs and physiological systems involved. These studies provide information for the guidance of the project team in making further plans, but are not normally part of the regulatory package that has to be approved before the drug can be given to humans, so they do not need to be perfomed under good laboratory practice (GLP) conditions.
• Regulatory toxicology. These studies are performed to GLP standards and comprise (a) those that are required by regulatory authorities, or by ethics committees, before the compound can be given for the first time to humans; (b) studies required to support an application for marketing approval, which are normally performed in parallel with clinical trials. Full reports of all studies of this kind are included in documentation submitted to the regulatory authorities.
The basic procedures for safety assessment of a single new synthetic compound are fairly standard, although the regulatory authorities have avoided applying a defined checklist of tests and criteria required for regulatory approval. Instead they issue guidance notes (available on relevant websites – USA: www.fda.gov/cder/; EU: www.emea.eu.int; international harmonization guidelines: www.ich.org), but the onus is on the pharmaceutical company to anticipate and exclude any unwanted effects based on the specific chemistry, pharmacology and intended therapeutic use of the compound in question. The regulatory authorities will often ask companies developing second or third entrants in a new class to perform tests based on findings from the class leading compound.
There are many types of new drug applications that do not fall into the standard category of synthetic small molecules, where the safety assessment standards are different. These include most biopharmaceuticals (see Chapter 12), as well as vaccines, cell and gene therapy products (see below). Drug combinations, and non-standard delivery systems and routes of administration are also examples of special cases where safety assessment requirements differ from those used for conventional drugs. These special cases are not discussed in detail here; Gad (2002) gives a comprehensive account.
Types of adverse drug effect
Adverse reactions in man are of four general types:
• Exaggerated pharmacological effects – sometimes referred to as hyperpharmacology or mechanism-related effects – are dose related and in general predictable on the basis of the principal pharmacological effect of the drug. Examples include hypoglycaemia caused by antidiabetic drugs, hypokalaemia induced by diuretics, immunosuppression in response to steroids, etc.
• Pharmacological effects associated with targets other than the principal one – covered by the general term side effects or off-target effects. Examples include hypotension produced by various antipsychotic drugs which block adrenoceptors as well as dopamine receptors (their principal target), and cardiac arrythmias associated with hERG-channel inhibition (see below). Many drugs inhibit one or more forms of cytochrome P450, and hence affect the metabolism of other drugs. Provided the pharmacological profile of the compound is known in sufficient detail, effects of this kind are also predictable (see also Chapter 10).
• Dose-related toxic effects that are unrelated to the intended pharmacological effects of the drug. Commonly such effects, which include toxic effects on liver, kidney, endocrine glands, immune cells and other systems, are produced not by the parent drug, but by chemically reactive metabolites. Examples include the gum hyperplasia produced by the antiepileptic drug phenytoin, hearing loss caused by aminoglycoside antibiotics, and peripheral neuropathy caused by thalidomide1. Genotoxicity and reproductive toxicity (see below) also fall into this category. Such adverse effects are not, in general, predictable from the pharmacological profile of the compound. It is well known that certain chemical structures are associated with toxicity, and so these will generally be eliminated early in the lead identification stage, sometimes in silico before any actual compounds are synthesized. The main function of toxicological studies in drug development is to detect dose-related toxic effects of an unpredictable nature.
• Rare, and sometimes serious, adverse effects, known as idiosyncratic reactions, that occur in certain individuals and are not dose related. Many examples have come to light among drugs that have entered clinical use, e.g. aplastic anaemia produced by chloramphenicol, anaphylactic responses to penicillin, oculomucocutaneous syndrome with practolol, bone marrow depression with clozapine. Toxicological tests in animals rarely reveal such effects, and because they may occur in only one in several thousand humans they are likely to remain undetected in clinical trials, coming to light only after the drug has been registered and given to thousands of patients. (The reaction to clozapine is an exception. It affects about 1% of patients and was detected in early clinical trials. The bone marrow effect, though potentially life-threatening, is reversible, and clozapine was successfully registered, with a condition that patients receiving it must be regularly monitored.)
Safety pharmacology
The pharmacological studies described in Chapter 11 are exploratory (i.e. surveying the effects of the compound with respect to selectivity against a wide range of possible targets) or hypothesis driven (checking whether the expected effects of the drug, based on its target selectivity, are actually produced). In contrast, safety pharmacology comprises a series of protocol-driven studies, aimed specifically at detecting possible undesirable or dangerous effects of exposure to the drug in therapeutic doses (see ICH Guideline S7A). The emphasis is on acute effects produced by single-dose administration, as distinct from toxicology studies, which focus mainly on the effects of chronic exposure. Safety pharmacology evaluation forms an important part of the dossier submitted to the regulatory authorities.
ICH Guideline S7A defines a core battery of safety pharmacology tests, and a series of follow-up and supplementary tests (Table 15.1). The core battery is normally performed on all compounds intended for systemic use. Where they are not appropriate (e.g. for preparations given topically) their omission has to be justified on the basis of information about the extent of systemic exposure that may occur when the drug is given by the intended route. Follow-up studies are required if the core battery of tests reveals effects whose mechanism needs to be determined. Supplementary tests need to be performed if the known chemistry or pharmacology of the compound gives any reason to expect that it may produce side effects (e.g. a compound with a thiazide-like structure should be tested for possible inhibition of insulin secretion, this being a known side effect of thiazide diuretics; similarly, an opioid needs to be tested for dependence liability and effects on gastrointestinal motility). Where there is a likelihood of significant drug interactions, this may also need to be tested as part of the supplementary programme.
Table 15.1 Safety pharmacology
| Type | Physiological system | Tests |
|---|---|---|
| Core battery | Central nervous system | Observations on conscious animals |
| Motor activity | ||
| Behavioural changes | ||
| Coordination | ||
| Reflex responses | ||
| Body temperature | ||
| Cardiovascular system | Measurements on anaesthetized animals | |
| Blood pressure | ||
| Heart rate | ||
| ECG changes | ||
| Tests for delayed ventricular repolarization (see text) | ||
| Respiratory system | Measurements on anaesthetized or conscious animals | |
| Respiratory rate | ||
| Tidal volume | ||
| Arterial oxygen saturation | ||
| Follow-up tests (examples) | Central nervous system | Tests on learning and memory |
| More complex test for changes in behaviour and motor function | ||
| Tests for visual and auditory function | ||
| Cardiovascular system | Cardiac output | |
| Ventricular contractility | ||
| Vascular resistance | ||
| Regional blood flow | ||
| Respiratory system | Airways resistance and compliance | |
| Pulmonary arterial pressure | ||
| Blood gases | ||
| Supplementary tests (examples) | Renal function | Urine volume, osmolality pH |
| Proteinuria | ||
| Blood urea/creatinine | ||
| Fluid/electrolyte balance | ||
| Urine cytology | ||
| Autonomic nervous system | Cardiovascular, gastrointestinal and respiratory system responses to agonists and stimulation of autonomic nerves | |
| Gastrointestinal system | Gastric secretion | |
| Gastric pH | ||
| Intestinal motility | ||
| Gastrointestinal transit time | ||
| Other systems (e.g. endocrine, blood coagulation, skeletal muscle function, etc.) | Tests designed to detect likely acute effects |
The core battery of tests listed in Table 15.1 focuses on acute effects on cardiovascular, respiratory and nervous systems, based on standard physiological measurements.
The follow-up and supplementary tests are less clearly defined, and the list given in Table 15.1 is neither prescriptive nor complete. It is the responsibility of the team to decide what tests are relevant and how the studies should be performed, and to justify these decisions in the submission to the regulatory authority.
Tests for QT interval prolongation
The ability of a number of therapeutically used drugs to cause a potentially fatal ventricular arrhythmia (’torsade de pointes’) has been a cause of major concern to clinicians and regulatory authorities (see Committee for Proprietary Medicinal Products, 1997; Haverkamp et al., 2000). The arrhythmia is associated with prolongation of the ventricular action potential (delayed ventricular repolarization), reflected in ECG recordings as prolongation of the QT interval. Drugs known to possess this serious risk, many of which have been withdrawn, include several tricyclic antidepressants, some antipsychotic drugs (e.g. thioridazine, droperidol), antidysrhythmic drugs (e.g. amiodarone, quinidine, disopyramide), antihistamines (terfenadine, astemizole) and certain antimalarial drugs (e.g. halofantrine). The main mechanism responsible appears to be inhibition of a potassium channel, termed the hERG channel, which plays a major role in terminating the ventricular action potential (Netzer et al., 2001).
Proposed standard tests for QT interval prolongation have been formulated as ICH Guideline S7B. They comprise (a) testing for inhibition of hERG channel currents in cell lines engineered to express the hERG gene; (b) measurements of action potential duration in myocardial cells from different parts of the heart in different species; and (c) measurements of QT interval in ECG recordings in conscious animals. These studies are usually carried out on ferrets or guinea pigs, as well as larger mammalian species, such as dog, rabbit, pig or monkey, in which hERG-like channels control ventricular repolarization, rather than in rat and mouse. In vivo tests for proarrhythmic effects in various species are being developed (De Clerck et al., 2002), but have not yet been evaluated for regulatory purposes.
Because of the importance of drug-induced QT prolongation in man, and the fact that many diverse groups of drugs appear to have this property, there is a need for high-throughput screening for hERG channel inhibition to be incorporated early in a drug discovery project. The above methods are not suitable for high-throughput screening, but alternative methods, such as inhibition of binding of labelled dofetilide (a potent hERG-channel blocker), or fluorimetric membrane potential assays on cell lines expressing these channels, can be used in high-throughput formats, as increasingly can automated patch clamp studies (see Chapter 8). It is important to note that binding and fluorescence assays are not seen as adequately predictive and cannot replace the patch clamp studies under the guidelines (ICH 7B). These assays are now becoming widely used as part of screening before selecting a clinical candidate molecule, though there is still a need to confirm presence or absence of QT prolongation in functional in vivo tests before advancing a compound into clinical development.
Exploratory (dose range-finding) toxicology studies
The first stage of toxicological evaluation usually takes the form of a dose range-finding study in a rodent and/or a non-rodent species. The species commonly used in toxicology are mice, rats, guinea pigs, hamsters, rabbits, dogs, minipigs and non-human primates. Usually two species (rat and mouse) are tested initially, but others may be used if there are reasons for thinking that the drug may exert species-specific effects. A single dose is given to each test animal, preferably by the intended route of administration in the clinic, and in a formulation shown by previous pharmacokinetic studies to produce satisfactory absorption and duration of action (see also Chapter 10). Generally, widely spaced doses (e.g. 10, 100, 1000 mg/kg) will be tested first, on groups of three to four rodents, and the animals will be observed over 14 days for obvious signs of toxicity. Alternatively, a dose escalation protocol may be used, in which each animal is treated with increasing doses of the drug at intervals (e.g. every 2 days) until signs of toxicity appear, or until a dose of 2000 mg/kg is reached. With either protocol, the animals are killed at the end of the experiment and autopsied to determine if any target organs are grossly affected. The results of such dose range-finding studies provide a rough estimate of the no toxic effect level (NTEL, see Toxicity measures, below) in the species tested, and the nature of the gross effects seen is often a useful pointer to the main target tissues and organs.
The dose range-finding study will normally be followed by a more detailed single-dose toxicological study in two or more species, the doses tested being chosen to span the estimated NTEL. Usually four or five doses will be tested, ranging from a dose in the expected therapeutic range to doses well above the estimated NTEL. A typical protocol for such an acute toxicity study is shown in Figure 15.2. The data collected consist of regular systematic assessment of the animals for a range of clinical signs on the basis of a standardized checklist, together with gross autopsy findings of animals dying during a 2-week observation period, or killed at the end. The main signs that are monitored are shown in Table 15.2.
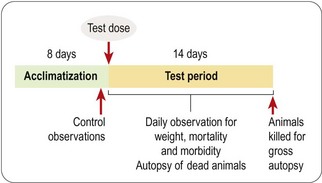
Table 15.2 Clinical observations in acute toxicity tests
| System | Observation | Signs of toxicity |
|---|---|---|
| Nervous system | Behaviour | Sedation |
| Restlessness | ||
| Aggression | ||
| Motor function | Twitch | |
| Tremor | ||
| Ataxia | ||
| Catatonia | ||
| Convulsions | ||
| Muscle rigidity or flaccidity | ||
| Sensory function | Excessive or diminished response to stimuli | |
| Respiratory | Respiration | Increased or decreased respiratory rate |
| Intermittent respiration | ||
| Dyspnoea | ||
| Cardiovascular | Cardiac palpation | Increase or decrease in rate or force |
| ?Electrocardiography | Disturbances of rhythm. | |
| Altered ECG pattern (e.g. QT prolongation) | ||
| Gastrointestinal | Faeces | Diarrhoea or constipation |
| Abnormal form or colour | ||
| Bleeding | ||
| Abdomen | Spasm or tenderness | |
| Genitourinary | Genitalia | Swelling, inflammation, discharge, bleeding |
| Skin and fur | Discoloration | |
| Lesions | ||
| Piloerection | ||
| Mouth | Discharge | |
| Congestion | ||
| Bleeding | ||
| Eye | Pupil size | Mydriasis or miosis |
| Eyelids | Ptosis, exophthalmos | |
| Movements | Nystagmus | |
| Cornea | Opacity | |
| General signs | Body weight | Weight loss |
| Body temperature | Increase or decrease |
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


