Articular (hyaline) cartilage is a tissue composed of collagen bundles intertwined with noncollagenous proteins and negatively charged proteoglycan molecules attracting water (Fig. 16.1). The collagen bundles consist of types II, XI, and IX in an 8:1:1 ratio. The proteoglycan acts like a gel within which the fibrils of collagen provide a three-dimensional reinforcement. The structure has both biologic and biomechanical functions. The collagen fibrils most likely serve to anchor the proteoglycan matrix protecting chondrocytes. In addition, they contribute to resisting extrinsic forces during loading and resisting the intrinsic swelling that occurs metabolically within the proteoglycan domain. Thus, collagen provides the tensile property of articular cartilage.
The main function of chondrocytes, which comprise only 10 percent of the volume of articular cartilage, is to produce and maintain the extracellular matrix, which consists of 65 to 80 percent water, 10 to 20 percent type II collagen, and aggrecan (complex proteoglycan molecules) (1).
By providing the swelling pressure necessary to resist compression, the compressive strength of articular cartilage is due to the proteoglycan aggrecan complexes.
Proteoglycans are complex glycoproteins consisting of multiple proteoglycan monomers attached to a hyaluronic acid backbone (hyaluronan) connected by a link protein aggrecan constituting >80 percent of the proteoglycan in articular cartilage. The proteoglycan monomer consists of a central protein core to which are attached repeated units of glycosaminoglycans (GAGs), sulfated disaccharides such as chondroitin-4-sulfate and chondroitin-6-sulfate (Fig. 16.1) (2). The early synthesis of these N-linked and O-linked oligosaccharides and their attachment to the precursor chondrocyte protein occur within the cytoplasmic membrane of the chondrocyte in the endoplasmic reticulum (3). Modifications of this structure occur in the Golgi. There is subsequent packaging into secretory vesicles and eventual secretion into the cartilage matrix.
The strong negative charges of the GAG molecules draw water into cartilage, giving it an ability to withstand compressive forces. These charges form the basis of novel imaging techniques to detect early cartilage damage.
Although standard magnetic resonance imaging (MRI) techniques can detect cartilage breakdown associated with morphologic changes such as alterations in cartilage volume and thickness, early biochemical changes in the cartilage matrix go undetected. The early diagnosis of cartilage injury from trauma or degenerative joint disease (DJD) requires the ability to discern subtle changes in collagen and proteoglycan integrity before gross and/or clinically serious damage has occurred. Techniques such as the exploitation of the charged ions in cartilage by use of the MRI anionic contrast agent gadolinium are imaging novelties to discern these early changes (4,5,6).
Aggrecan molecules, the aggregating proteoglycans that interact with the backbone hyaluronic acid, should be distinguished from nonaggregating proteoglycans, which do not interact with hyaluronic acid. Nonaggregating proteoglycans, such as decorin, biglycan, fibromodulin, and lumican, are much smaller than aggrecan, possess fewer dermatan sulfate and keratin sulfate chains, and probably function in collagen fibril interaction.
The mucopolysaccharidoses are examples of the profound effects on the skeleton due to disorders in which GAGs are not degraded by proteinases or intracellularly by lysosomal actions of enzymes such as sulfatases resulting in their intralysosomal accumulation (7,8) (Table 16.1, Fig. 16.2).
FIGURE 16.1. Articular cartilage: microarchitecture and aggregan. HA, hyaluronic acid; KS, keratan-sulfate chains; CS, chondroitin-sulfate chains; G1, binding region of aggrecan; G3, C-terminal end; COMP, cartilage oligomeric matrix protein; HS-PG, heparin sulfate proteoglycan; PRELP, proline arginine-rich end leucine-rich repeat protein.
TABLE 16.1 Summary of General and Musculoskeletal Features of the MPS Syndromes
MPS Type
Enzyme Deficiency
General Features
Musculoskeletal Features
MPS I (Hurler/Scheie)
α-L-iduronidase
Coarse facies, gum hypertrophy, macroglossia, initial intellectual development normal, then deteriorates, cardiomyopathy, aortic and mitral valve disease, hydrocephalus, hepatosplenomegaly
From Thakur AR, Naikmasur VG, Sattur A. Hurler syndrome: orofacial, dental, and skeletal findings of a case. Skeletal Radiol. 2015;;44(4):579-586. doi: 10.1007/s00256-014-1982-7.
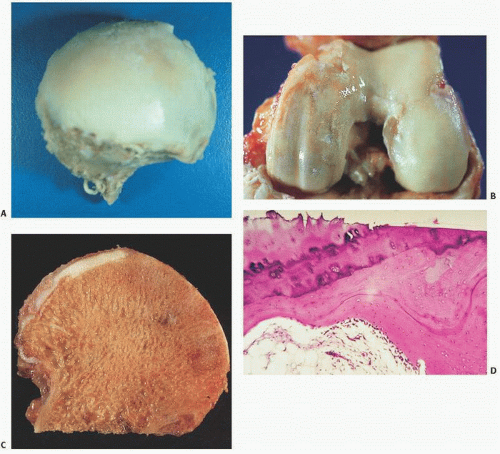
FIGURE 16.2. In the group of diseases known as mucopolysaccharidoses, glycosaminoglycans (GAGs) accumulate and osteoarticular changes, often apparent on roentgenogram, ensue. In Morquio disease, a deficiency of lysosomal enzymes causes GAG degradation to be blocked. The radiographic presentation of Morquio disease is similar to that of a multiple epiphyseal dysplasia. The epiphyses of the long bones are irregular. The irregularity in the ossification of the femoral heads can lead to confusion with avascular necrosis and Legg-Perthes-Calvés disease (A). The metaphyses tend to be widened and deformed. The diaphyses, in general, tend to have normal appearance. The pelvis, normal at birth, is narrow and elongated. The radiographic appearance of the pelvis has been compared with the contour of a wine glass. The vertebral bodies are flattened (platyspondyly) (B). The vertebral bodies have deformed contour, with an anterior bony projection between the upper and lower end plates. This central anterior tongue can be helpful in differentiating Morquio from Hurler disease, in which case the anterior tongue is located close to the upper end plate.
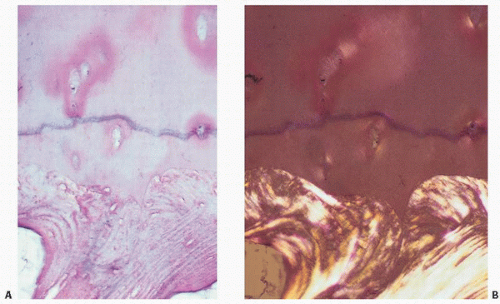
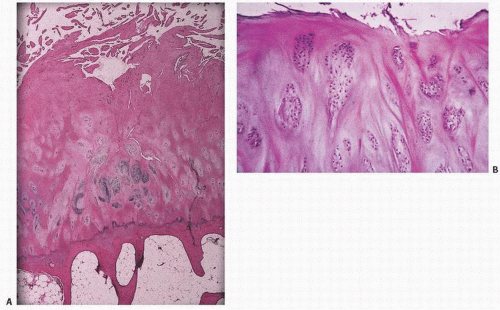
In general, articular cartilage can be divided into different zones based on the orientation of the collagen fibrils and chondrocytes. The layer immediately adjacent to the joint space consists of a fibrous surface that is in contact with the synovial fluid (Fig. 16.3). This superficial or tangential layer is often referred to as the “lamina splendens” and is relatively acellular, consisting mostly of collagen (9). In the superficial layer, the collagen fibers in long axes of the cells are parallel to the surface. The surface tissue is rich in collagen and has tightly bound smaller proteoglycans such as decorin and biglycan. There is much less of the large aggregating proteoglycans (aggrecan). Beneath this layer rests the intermediate or transitional zone followed by the basal or radial zone where tissue is rich in aggrecan and low in collagen. Chondrocytes are lined in columns similar to that seen at the growth plate (Fig. 16.4). At the deep end of the radial zone, the cartilage undergoes calcification at a distinct interface, which is recognizable by its basophilic properties on routine hematoxylin-eosin staining. This curvy, linear three-dimensional undulating interface is referred to as the “tidemark” or “mineralization front” (Fig. 16.5). Beneath this is a zone of calcified cartilage that is undulating in its width but often indistinct at touching points with the underlying subchondral bone (Fig. 16.5). The interface between the calcified cartilage and the subchondral bone is that of a layer of bone oriented in a platelike fashion as though the articular cartilage is resting on a platform (Fig. 16.6). The various zones of articular cartilage are somewhat arbitrarily defined as there are slight variations based on the diversity of the cartilage, the appearance and orientation of the chondrocytes, the distribution of collagen fibers, and other characteristics. In the most superficial layer of articular cartilage, which is approximately 200 µm in thickness, there is essentially little proteoglycan synthesized, and it consists mostly of fibrous tissue. The orientation of the fibrous cartilage tissue in the superficial zone is important in the biomechanical integrity of the articular cartilage. If stressed against its fiber orientation, collagen fibril breakdown can occur and may be the initial event in the development of DJD.
Although cartilage is predominantly composed of type II collagen, type VI is present in the superficial surface, and type X in the calcifying zone.
Normally, cartilage is avascular with the cartilage cells in the superficial zones deriving nutrition from the synovial fluid. More basal zones most likely obtain nutrition from subchondral bone.
Cartilage also lacks a neural structure.
Cartilage Metabolism
Articular cartilage is sparsely cellular, with cartilage cells lacking cell-to-cell contact. Diffusion of nutrients and metabolites through the matrix probably compensates for the lack of discernible blood and lymphatic tissue. Normally, cartilage matrix is metabolized and probably degradation products released into the synovial fluid. Collagen is turned over more slowly and is subject to the degradative collagenous enzymes. The proteoglycan breakdown is probably controlled by metalloproteinases secreted by chondrocytes. Innate to the normal physiology is the interaction between the relatively solid components of collagen proteoglycan and noncollagenous proteins and glycoproteins and the largely fluid phase consisting of essentially water and dissolved in organic salts. Biomechanically, it probably responds by both deformation of the solid matrix and redistribution of fluid as a response to compressive stresses. The interaction of the various matrix components based on electronic charge is most likely significant. In this regard, the differences in the articular cartilage between the normal and degenerative or osteoarthritic cartilage are of note.
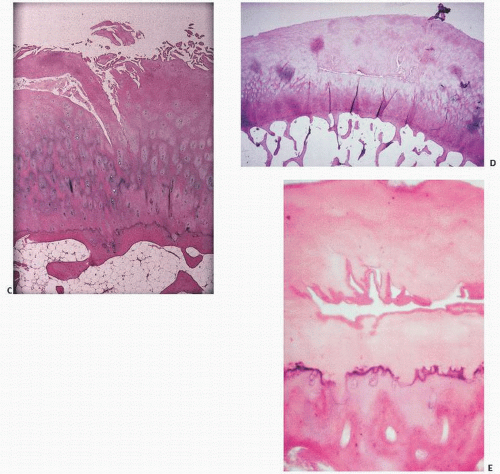
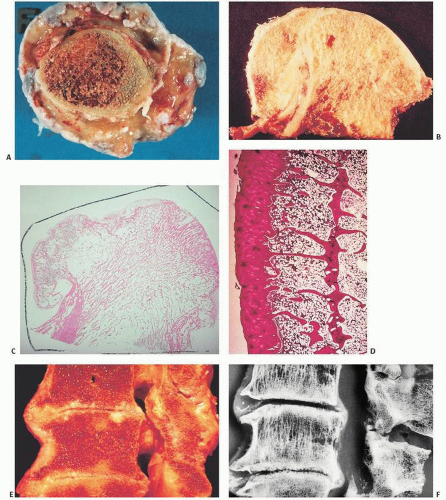
FIGURE 16.3. Articular cartilage composite. (A) Drawing of femur with insets showing the faint bluish-tinged gross appearance of youthful articular cartilage (left) versus the yellowish aging articular cartilage of a 70-year-old individual (right). (B) Coronal section of bone showing at its upper end the articular cartilage. Histologically, articular cartilage covers the end of the bone, its upper surface exposed to the joint space and its lower surface anchored to the bone (boxed area). Polarized microscopy demonstrates the horizontal arrangement of collagen at the joint interface and the subchondral bone below. (C) The interwoven architecture of collagen is seen arcading in vertically oriented bands within the cartilage and horizontally at its surface (right). (D) Light (left) and polarized light microscopy (right) of articular cartilage showing the horizontal orientation of chondrocytes and collagen at the joint surfaces, the “lamina splendens.”
Finally, in addition to the interactions of cartilage components based on the state of electrical charge, numerous growth factors and local tissue factors that are of importance in other connective tissue metabolism are of import here. Essentially, there are stimulatory effects of local growth factors such as tissue growth factor-b, fibroblast growth factor, and platelet-derived growth factor and bone morphogenetic proteins (10).
FIGURE 16.4. Most chondrocytes in articular cartilage are arranged in vertically oriented columns, not unlike the chondrocyte growth pattern at the growth plate.
FIGURE 16.5. The tidemark or mineralization front, identifiable on routine hematoxylin-eosin histologic sections as a slightly undulating basophilic line, is the point at which cartilage becomes calcified (A). The cartilage, calcified cartilage (the zone between the tidemark and bone), and subchondral bone are most apparent utilizing polarized light (B).
Degenerative Joint Disease (Osteoarthritis)
Degenerative joint disease (osteoarthritis [OA]) is the most common disease involving diarthrodial joints. In 2005, in the United States the number of people with DJD was estimated at 12 percent of the U.S. population, affecting a staggering 27 million people and an annual health-care cost of approximately 90 billion dollars. When symptomatic, which is not always the case, DJD causes pain, stiffness, and functional disability. Although the etiology of OA (DJD) is unknown, OA remains a convenient term for a broad range of both local and systemic disorders that have in common subtle synovial changes, alteration, and eventual loss of articular cartilage, and roentgenographically detectable narrowing of the joint space and remodeling of bone (3,11) (Table 16.2). Although inflammation may be seen microscopically in joint tissue removed at surgery, the amount is minimal in most cases, and therefore the term osteoarthritis is misleading. The prototype of inflammatory arthritis is rheumatoid arthritis (RA).
To explain the myriad clinical situations in which DJD can develop, a multifactorial disease hypothesis is most attractive. Most likely, the disorder is caused by an interplay of systemic risk factors (e.g., age, gender, hormone levels, genetic predisposition, metabolic disease such as ochronosis, hemochromatosis, and nutrition), intrinsic joint factors (e.g., anatomic variants such as dysplasias, muscle weakness, malalignment, and joint laxity), and extrinsic risk factors (e.g., excessive joint loading as seen in obesity and repetitive physical activities).
There is little doubt that the whole joint is involved in DJD. Cartilage degenerates, bone remodels at an accelerated rate, forming sclerosis and cysts. Bone marrow changes include fibrosis, edema, trabecular bone microfractures, and foci of necrosis. Tendons and ligaments (rarely studied) show degenerative changes, and the synovium undergoes transient inflammatory and metaplastic changes to cartilage and bone (12).
It is widely believed that the integrity of a joint requires an even distribution of load across the joint. Therefore, resiliency of the subchondral bone, and proper alignment and normal biologic integrity of associated joint structures are essential. Abnormalities of alignment (e.g., trauma, acromegaly), subchondral bone (e.g., Paget disease), and integrity of associated tissues (e.g., ochronosis, calcium pyrophosphate crystal deposition diseases) may predispose to DJD. In addition, whereas mechanical factors such as obesity have been considered protective against the development of osteoporosis, an association between OA of the knee and obesity has been established (Fig. 16.7).
FIGURE 16.6. At the subchondral bone zone, pockets of woven bone can be seen above mature lamellar bone (bottom).
FIGURE 16.7. Factors contributing to the evolution of osteoarthritis. (After Felson DT. Risk factors for osteo arthritis. Understanding joint vulnerabilities. Clin Orthop. 2004;427 (suppl):S16-S21, with permission.)
Could DJD be the inevitable consequence of evolution? Consider femoroacetabular impingement (FAI). From an evolutionary perspective, FAI may actually be the norm. Rarely found in animals that climb and swim, coax recta, the animal equivalent of FAI, is commonly found in species that run and jump (13).
TABLE 16.2. Classification of Degenerative Joint Disease (Osteoarthritis)
A commonly held belief is that “wear and tear” on the joint predisposes one to the development of DJD. Of note are the following:
Obesity is definitely linked to DJD of the knee.
Meniscectomy leads to symptomatic osteoarthritis of the knee with a 132-fold increase in the rate of total knee replacement compared with geographically and age-matched peers (14).
Ligament injuries increase the risk of DJD 10-fold.
Roughly 12 percent of DJD is posttraumatic, and the pathogenetic link is most likely the release of prostaglandin E2, interleukin 6, and nitric oxide. Impact trauma results in articular cartilage damage via chondrocyte death and disruption of the extracellular matrix.
Alternative to (and not necessarily exclusive of) the mechanical hypothesis for the etiology of OA is the possibility that change of the articular cartilage de novo precipitates the degenerative process. In this regard, distinction from the normal aging process of cartilage is instructive. Cartilage is mostly composed of water plus proteoglycans (GAGs, core protein, and oligosaccharides), type II collagen, and glycoproteins such as chondronectin and fibronectin that promote interaction with chondrocytes to form the peculiar characteristics of the cartilage matrix—that is, an ability to retain water within an organized collagenous mesh. Both aging and DJD have been associated with changes (albeit different) in water content and proteoglycan type, size, and aggregation (3) (Table 16.3).
TABLE 16.3 Comparison of Changes in Articular Cartilage in Aging and Osteoarthritis
Criterion
Aging
Osteoarthritis
Gross
Cartilage color
Yellowing
No pigment changes initially
Microscopic
Fibrillation
Non-weight-bearing surfaces
Often affects weight-bearing joints
No cleft formation
Present with clefting and slits
Minimal physical and chemical changes
Significant physical and chemical changes
Eburnation
No
Yes Osteophytes occur with cartilage and subchondral bone changes. New bone forms where muscle and capsular stresses are increased.
Tissue volume
No increase
Almost all periarticular and intra-articular tissues are markedly increased.
Biochemical
Water content
Decreased
Increased
Glycosaminoglycans
Chondroitin surface
Normal or slightly less
Decreased
Chondroitin sulfate 4/6 ratio
Decreased
Increased
Keratan sulfate
Increased
Decreased
Hyaluronate
Increased
Decreased
Proteoglycans
Aggregation
Normal
Diminished
Monomer size
Decreased
Decreased
Link protein
Fragmented
Normal
Proteases
“Normal”
Increased
Collagen
Increased cross-linking
Disruption of collagen organization
Increased fiber diameters
Variable fiber diameters
Modified from Hammerman D. The biology of osteoarthritis. N Engl J Med. 1989;230:1322-1330.
A major age-related change in articular cartilage is the spontaneous modification of proteins by nonenzymatic glycation, resulting in the accumulation of advanced glycation end products called AGEs. The accumulation of AGEs has been correlated with increased tissue stiffness of cartilage.
In DJD, these changes have been postulated to lead to water retention and proteoglycan dilution and subsequent loss, perhaps by diffusion. Eventually, the ability to withstand transarthrodial forces decreases. In addition, cells may produce and release products, such as cytokines (e.g., interleukin), that can cleave proteoglycan units and therefore cause them to deteriorate. Thus, early DJD is characterized by a depletion of proteoglycans and loosening of the collagen network. A role for the synovium is not excluded in this scenario, as the synovium may directly produce damaging cytokines perhaps by processing the degrading and degenerating cartilage fragments.
The classic findings on routine radiographic imaging of DJD such as narrowing of the joint space, subchondral cysts, osteophytes, and loose bodies occur late in the disorder, and so there remains a need to diagnose the disorder at a much earlier stage.
Bone marrow edema-like lesions (BMELs), defined as regions with increased signal intensity in areas of subchondral bone marrow in fluid-sensitive sequences (such as fat-saturated T2-weighted fast spin-echo sequences or short tau inversion recovery [STIR] sequences in MRI), are thought to be more prevalent in patients with knee OA and may represent a number of histological abnormalities, including bone marrow necrosis, bone marrow fibrosis, trabecular abnormalities, and a small amount of edema in osteoarthritic knees (15).
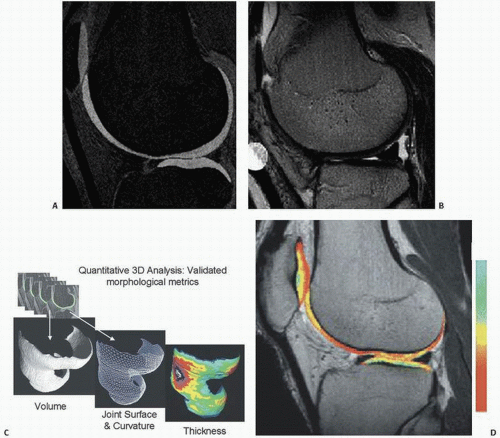
The continuing attempt to diagnose DJD at an early stage has led to novel imaging techniques exploiting the negative charges in the GAG component of articular cartilage (Fig. 16.8).
Since loss of cartilage GAG is thought to be one of the earliest reversible changes of cartilage degeneration and, thus, early DJD, techniques to explore this change have been studied. One such noninvasive technique is delayed gadolinium-enhanced MR imaging of cartilage (dGEMRIC). In articular cartilage, the distribution of ions within the interstitial fluid of cartilage is dependent on the fixed charge density induced by the negatively charged GAG. Since the distribution of an anionic contrast agent such as Gd-DTPA2 given sufficient time to reach equilibrium, is inversely proportional to the fixed negative charge of GAG, a higher concentration of Gd-DTPA2, is seen in areas with low GAG, and vice versa in accordance with the Donnan principle of electroneutrality (16). The T1 measurement after penetration of Gd-DTPA2 is referred to as the dGEMRIC index.
Theories for the pathogenesis of DJD can be summarized as those initiating in cartilage, subchondral bone, or the synovium. In cartilage, a change in the microenvironment of chondrocytes may be the culprit (3) (Fig. 16.7).
Alternatively, the mechanical factors affecting or affected by subchondral bone may precipitate DJD (11). Here, fatigue failure and microfractures leading to altered biomechanics and eventually increased stiffness are pathogenic possibilities. The alteration of subchondral bone by Paget disease is a most likely example.
Synovial tissue may also be activated by a broad range of both clinical and subclinical insults. Once activated, synovium may digest protein cores of proteoglycan moieties.
Other possibilities include microvascular disease with associated bone damage resulting from secondary micro-osteonecrosis. It is likely that the origin of DJD is multifactorial and that in any given clinical situation, alterations and/or damage to synovium, cartilage, and/or bone are contributory.
Some clinical observations are provocative and well established. OA increases with age, and although age-related changes overlap, they, unlike OA, are probably progressive. Deposited crystals (apatite or calcium pyrophosphate) are more likely markers than etiologic events. The natural history of OA is highly variable. Patients can improve, remain stable, or progressively worsen. Unlike RA, with its slow, inevitable progression, OA may stabilize and then remain unchanged or even regress focally.
The pathologic changes associated with these mechanical and biochemical processes (17) (Table 16.4) are evident during both gross and microscopic review of tissue removed at various stages of the degenerative process. The heterogeneity of DJD is appreciated when it is classified according to the clinical entities that precede it (Table 16.2). Numerous clinical variants have been proposed (Table 16.5).
For example, endemic types of DJD have been well documented such as Kashin-Beck disease, an osteoarthropathic disease of unknown cause. Endemic in parts of China, Russia, and North Korea but rare in the United States, the disease is most likely caused by a biological factor from a specific environmental origin such as the T-2 toxin contamination in food and selenium deficiency in the environment.
There are no laboratory tests that can diagnose OA and no biomarkers of the disease in regular clinical use.
Biomarkers under investigation include those that measure type II collagen turnover such as C-propeptide (CPII). Noncollagenous biomarkers under investigation include keratin sulfate, hyaluronic acid, and cartilage oligomeric matrix protein (COMP), all likely candidates as they are significant components of articular cartilage (18).
Tissue Changes
Tissue changes observed microscopically in DJD are mostly nonspecific but characteristic.
Synovium
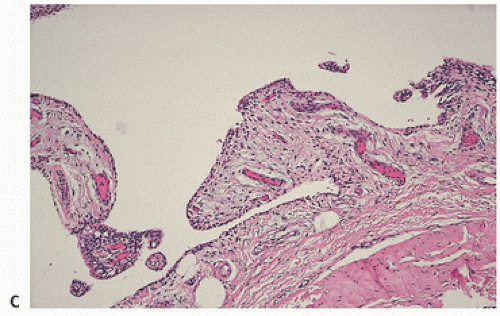
Normally, the synovium consists of a discrete layer of intimal lining cells, beneath which lies a richly vascular layer of loose connective tissue. In the osteoarthritic joint, the synovium becomes variably hyperplastic, often with increased villous folds and villous hypertrophy (Fig. 16.9). Microscopically, there is little discernible change. Hyperplasia and/or hypertrophy of the synovial lining cells may occur focally or diffusely. Inflammation is sparse but may be present, never to the extent seen in RA. Rarely acute, it usually consists of scattered mononuclear cells, including lymphocytes.
There is no doubt that some degree of synovitis is present in various stages of the disease and may be associated with greater levels of pain and joint symptoms (19). This is most probably the case in posttraumatic DJD. Variants of DJD with more marked inflammation are well described.
Osseocartilaginous loose bodies often form in the joint in association with DJD. Although other mechanisms may contribute, they are usually the result of synovial metaplasia (Fig. 16.10).
Articular Cartilage
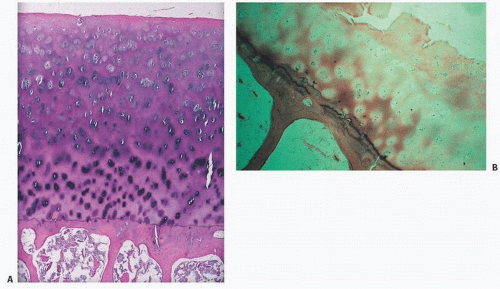
Normally, an organized structure with an even spatial distribution of chondrocytes, articular cartilage is avascular. It is sparsely cellular at its joint surface (the lamina splendens), with increasing cellularity toward the subchondral bone (Fig. 16.11). The site of calcification is identified as a basophilic wavy junction, the tidemark or mineralization front at the base of the articular cartilage (Fig. 16.12).
FIGURE 16.8. New imaging advances to detect early DJD. (A) T1-weighted spoiled gradient echo (SPGR) with fat suppression. (B) T2-weighted fast spin echo (FSE) with fat suppression. (C) Three-dimensional data for quantifying cartilage volume, joint morphology, and cartilage thickness. (D) An example of dGEMRIC image of the knee is shown, in which cartilage tissue has been colorized such that the color corresponds with the relative amount of negative charge or glycosaminoglycan (GAG), with red being low, and yellow being high. Healthy cartilage with abundant glycosaminoglycan is colored yellow. As such, the dGEMRIC image provides molecular image of cartilage by MRI. (After Gray ML, Eckstein F, Peterfy C, et al. Toward imaging biomarkers for osteoarthritis. Clin Orthop. 2004;427 (suppl):175-181, with permission.)
The main pathophysiological changes in DJD is the breakdown of proteoglycans and collagen fibers caused by the increased activity of matrix metalloproteinase (MMPs) and members of the ADAMTS (a disintegrin and metalloproteinase with thrombospondin motif) family. Initially edematous, early cartilage lesions are primarily characterized by proteoglycan depletion and loosening of the collagen framework, which may reduce the osmotic pressure within the matrix. Cartilage becomes less resilient, unable to recover with hydration following the release of compression. Collagen degradation impairs resistance to compressive stress.
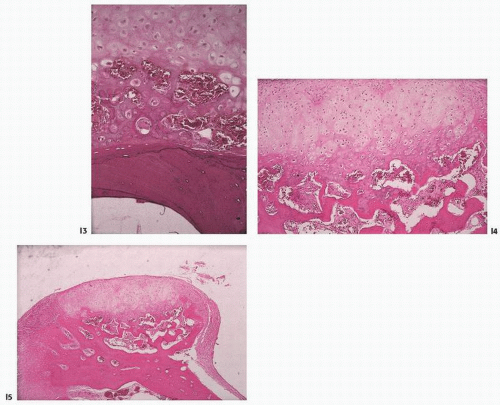
Changes seen in tissue removed during surgery for symptomatic DJD include unevenness and fibrillation or fraying of the surface (Fig. 16.13). The cartilage becomes thin, and fissures, clefts, or slits form (Fig. 16.14). Fissures are both vertical and horizontal in orientation. Often, chondrocytes proliferate adjacent to the fissures. These cartilage clones, or brood capsules (Fig. 16.15), may represent, at various stages in the evolution of DJD, reparative or degenerative manifestations of the disease. They are often seen adjacent to fibrillated or fraying cartilage.
TABLE 16.4 Selected Skeletal Biochemical Events in Osteoarthritis
Mechanical
Increased hydration (altered network of collagen fibers) leads to dilution and thus loss (by diffusion) of proteoglycans. Mechanical stresses produce more deformation and increased pressure on subchondral bone.
Biochemical
Alterations in proteoglycan size and aggregation
Decreased proteoglycan content
Alterations in collagen fibril size and weave
Decrease in keratan sulfate
Increase in sulfate ester on 4/6 carbon
Decrease in hyaluronic acid
Increased neutral metalloproteases
Disruption of collagen organization
Traces of type I collagen and glycosylated type II collagen
Increased arachidonic acid (precursor of prostaglandins, which in turn affect chondrocytes)
Prostaglandin E2 stimulation of cartilage to synthesize cyclic AMP, leading to production of lysosomal enzymes
Cathepsin-like protease, lysozyme, hyaluronidase, and aryl sulfatase degradation of proteoglycans
Interleukin-1 and lysosomal enzyme-triggered joint destruction by hydrolytic factor release from synovial membrane cells and chondrocytes, which degrades proteoglycan and collagen.
Modified after Hamerman D, Klagsbrun M. Osteoarthritis. Emerging evidence for cell interactions in the breakdown and remodeling of cartilage. Am J Med. 1985;78:495-499 and Kuettner KE, Peyron JG, Schleyerbach R, et al., eds. Articular Cartilage and Osteoarthritis: Workshop Conference. Hoechst Werk Kalle-Albert, Wiesbaden. New York, NY: Raven Press; 1992.
TABLE 16.5 Clinical Variants of Degenerative Joint Disease (DJD)
Generalized osteoarthritis
Rapidly destructive DJD
Erosive DJD
Crystal deposition, especially
Calcium pyrophosphate deposition disease
Hydroxyapatite arthropathy
Diffuse idiopathic skeletal hyperostosis
DJD, degenerative joint disease.
FIGURE 16.9. Synovium in degenerative joint disease (gross and microscopic). Normally, synovial tissue microscopically consists of a fine layer of synovial intimal cells resting on a fibrovascular zone (A). (Continued)
The term chondron refers to the entire chondrocyte complex consisting of the chondrocyte, its lacunar space, and its pericellular rim. As a metabolic entity, it is poorly understood.
Microscopically, OA is characterized by a marked variation in cellularity and variation in intensity of the proteoglycan matrix, which can be identified by special stains such as alcian blue and safranin O (Fig. 16.16).
Absence or loss of articular cartilage staining by stains such as toluidine blue or safranin O correlates with loss or absence of glycosaminoglycans, and is considered by many to be pathognomonic for arthritis.
FIGURE 16.9. (Continued) In degenerative joint disease, there is moderate proliferation of the synovium into villous projections [(B) gross; (C) microscopic].
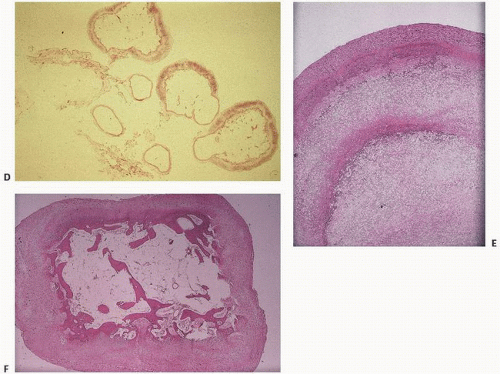
FIGURE 16.10. Composite. Synovial osseocartilaginous metaplasia (loose bodies) in degenerative joint disease. Roentgenogram of a knee with degenerative joint disease and secondary osseocartilaginous loose body formation (A). Loose bodies in degenerative joint disease form primarily by synovial metaplasia to cartilage, which can then form bone by endochondral ossification (B, C). (Continued)
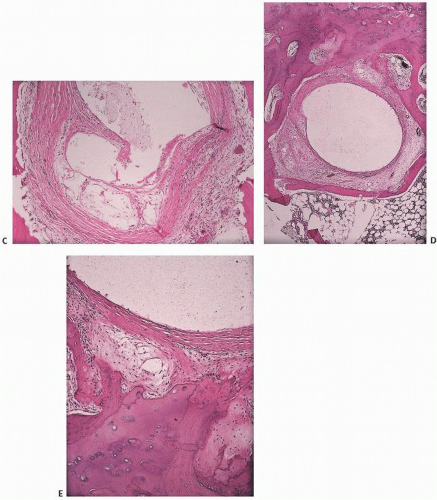
At the base of the articular cartilage, vascular penetration is noted from the underlying subchondral bone, with duplication and marked irregularity, to the tidemark (Fig. 16.17).
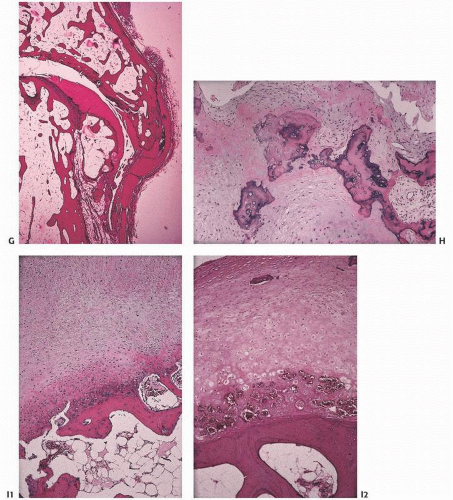
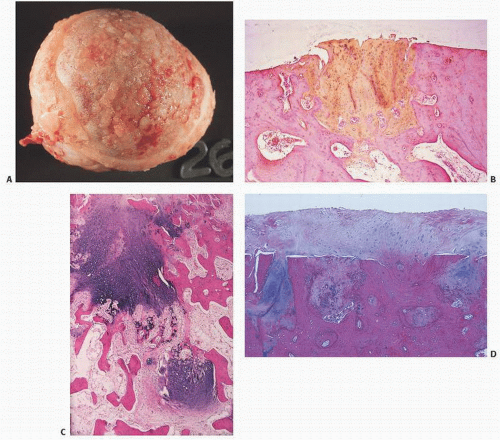
Eventually, the cartilage is denuded (eburnation) (Fig. 16.18). Mesenchymal proliferation ensues, and endochondral ossification similar to that seen at the growth plate results eventually in a discernible region of new articular bone—the osteophyte (Fig. 16.19).
Osteophytes
Osteophytes are growths of new bone, classically seen in the osteoarthritic joint. Usually found at the margins of osteoarthritic cartilage in nonpressure zones (20), they form by endochondral ossification (Fig. 16.19).
Radiographically, osteophytes appear as new lips or spurs of bone around the edges of a joint. They vary considerably in size with large proboscis-like osteophytes characteristic of acromegalic degenerative joint disease.
Osteophytes are capped by a more fibrocartilaginous tissue than the articular cartilage, a distinction that is most noted in ochronosis, where there is no pigment deposition (Vide infra, see Fig. 16.52C).
FIGURE 16.10. (Continued) Synovium in degenerative joint disease (gross and microscopic). Here, the synovium is transformed into an osseous loose body, which on cross section reveals bone. Loose bodies may consist of varying amounts of cartilage and bone (D). Typical loose bodies in degenerative joint disease show concentric rings of calcification (E) or abrupt transformation of fibrous or cartilaginous tissue into bone (F).
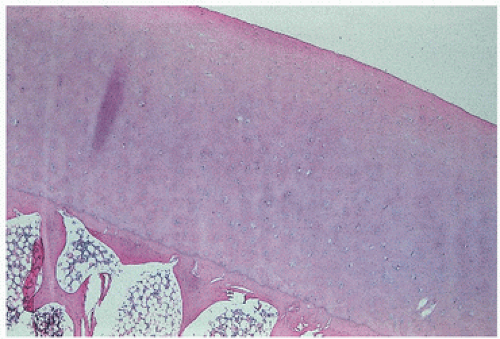
FIGURE 16.11. Normal articular cartilage demonstrates an even thickness with sparse cellularity at its surface (the lamina splendens). There is increased cellularity toward subchondral bone. The tissue is avascular.
FIGURE 16.12. Mineralization takes place at the base of articular cartilage at the mineralization front, or “tidemark.” Normally, the tidemark is a discrete solitary line, with clearly demarcated cartilage above and calcified cartilage and subchondral bone below [hematoxylin-eosin, routine light microscopy (left), phase-contrast microscopy (right)].
Often found near tendon insertions or capsular attachments, they form at the junction of hyaline cartilage and synovium or periosteum.
Regarded as an attempt at repair or redistribution of abnormal joint loading, their origin is obscure. Metaplasia of local synovium into cartilage or the result of migration and transformation of pluripotential mesenchymal cells are attractive hypotheses.
Osteophytes can arise in unusual sites such as the intercondylar notch of the knee, causing confusion with loose bodies. Central osteophytes are sometimes referred to as “stud” or “button” osteophytes, and those along the femoral neck “buttressing” osteophytes.
Osteophyte cartilage has been shown to be different than normal articular cartilage. Osteophytic and articular chondrocytes significantly differ in their gene expression. Articular cartilage expresses inhibitors of the BMP- and WNT-pathway, which may serve to stabilize permanent chondrocyte phenotype and thus prevent terminal differentiation. In contrast, osteophytic chondrocytes express genes involved in the endochondral ossification process (20).
FIGURE 16.13. The progression, grading, and imaging of osteoarthritis.
FIGURE 16.14. Marked fibrillation of articular surface in degenerative joint disease [(A) gross; (B) microscopic]. (Continued)
FIGURE 16.14. (Continued) Fissures and clefts in degenerative joint disease are either horizontally oriented (C), or vertical (D, E), disrupting the matrix components of cartilage.
Subchondral Bone
Although a hallmark of DJD is a progressive degradation of articular cartilage, changes in the subchondral bone are significant and include:
In addition, subchondral DJD osteoblasts express more factors such as IL-6, adipokines, and VEGF, suggesting a heightened metabolic state (22).
To what extent this alters bone compliance remains speculative. The contribution of subchondral bone in Paget disease to the development of DJD in that disorder suggests that subchondral bone is a major player in the evolution of osteoarthritis.
Scoring systems have been devised to grade the severity of histologic changes seen in association with DJD (Table 16.6).
Fibrocartilage
DJD affects fibrocartilaginous tissue and ligaments and tendons as well. Fibrocartilage ligament changes in DJD include myxoid degeneration, microcyst formation, chondroid metaplasia, and even deposition of calcium pyrophosphate dihydrate crystals. DiFrancesco and Sokoloff (23) described a “lipochondral” degeneration characterized by nodular chondroid metaplasia with vacuolar distension of chondrocytes, neurobiosis, and acellular pools of lipid that corresponded grossly to canary yellow material.
FIGURE 16.15. (A) Cartilage clones, or “brood capsules,” in degenerative joint disease. (B) Higher power.
FIGURE 16.16. Accentuation of matrix-staining variability and cellularity in early stages of degenerative joint disease.
Repair
Once lost, articular cartilage has limited regenerative capacity. However, the new tissue is more fibrous. It derives from subchondral tissue and may actually resurface parts of the joint (Fig. 16.21).
FIGURE 16.17. Articular bone interface in degenerative joint disease showing duplication of the tidemark and penetration of vascular cones into the calcified cartilage zone beneath the tidemark.
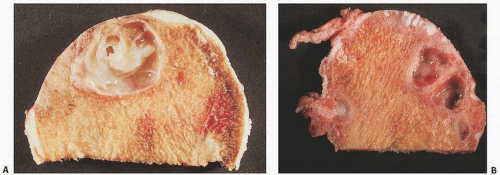
FIGURE 16.18. Progressive loss of articular cartilage from a femoral head (A) and the left femoral condyle of the knee (B). In (B), the left condyle shows complete eburnation. Complete loss of articular cartilage leaves denuded and exposed eburnated, or polished, subchondral bone. (C) Coronal section of the hip in a gross specimen, left surface intact, right surface completely eburnated. (D) Microscopic appearance.
On the basis of numerous histologic observations, scoring systems have been proposed (24,25,26,27,28) (Table 16.6). This sequence of pathologic changes can be formulated in the developing osteoarthritic joint.
Synovium becomes hyperplastic but nonspecifically so. Fluid analysis may reveal increased activity of collagenases and lysosomal or nonlysosomal enzymes. Subtle remodeling of subchondral bone, vascular penetration into subchondral bone near the tidemark (29), and irregularity or duplication of the tidemark (30) are seen. In the cartilage, collagen fibers begin to change their anatomic orientation, chondrocytes increase in number, and proteoglycan synthesis is decreased.
Later, fibrillation in cartilage is apparent, with cartilage thinning and further chondrocyte cloning and proliferation. Cell proliferation is accompanied by a paradoxical decrease in overall proteoglycan matrix production. Horizontal splitting at the interface between calcified and uncalcified cartilage may occur.
Late changes include total loss of articular cartilage with exposure of underlying bone (eburnation). Osteophytes form at the edges of the articular bone, and cysts form within the subchondral bone.
FIGURE 16.19. DJD. Osteophytes form when well-vascularized mesenchymal tissue at articular margins undergoes endochondral ossification. New cartilage, bone, and even marrow forms. In a view directed upward toward a femoral head, the overhanging new cartilage (white) and bone are readily apparent (A). A coronal section through a femoral head shows a large osteophyte (left) that has been added to the now completely eburnated and remodeled head [(B) gross; (C) histology; (D) osteophyte histology]. Formation of an osteophyte. Osteophytes commonly occur in spinal degenerative joint disease, and appear either as lips at the margins of the vertebral end plates (left) or as remodeling of posterior elements [(E) gross specimen; (F) x-ray film]. (Continued)
FIGURE 16.19. (Continued) Osteophytes at interphalangeal joints are referred to clinically as Heberden nodes if distal, and Bouchard nodes if proximal. New bone formation may be orderly (G) or disorganized (H). In (I1) and (I2), an osteophyte is evolving. (Continued)
Recently, the role of cytokines, proteases, and growth factors in the pathogenesis of OA has received considerable attention (31,32) (Table 16.4). Of these, interleukin-1b has been identified as a proinflammatory cytokine that may induce the breakdown of proteoglycans. It stimulates the synthesis of metalloproteases and plasminogen activator, inhibits proteoglycan synthesis, and decreases the synthesis of type II and type XI collagen.
Tumor necrosis factor-a, a cytokine, may help produce and be synergistic with interleukin-1b. Stromelysin, a metalloprotease, degrades proteoglycans and collagen and is activated by plasminogen activator.
Fibronectin, an extracellular glycoprotein, binds cells, collagen, and proteoglycans. It is greatly elevated in osteoarthritic cartilage.
Roentgenographic features of OA are well described (Fig. 16.22). Interestingly, before routine changes appear on x-ray films, bone scans have shown abnormal uptake in sites with OA that eventually becomes clinically evident, supporting bone remodeling as a significant etiologic factor.
FIGURE 16.19. (Continued) In (I3-I5), the osteophyte continues to evolve. Cartilage forms at the rim of eburnated bone and undergoes endochondral ossification.
FIGURE 16.20. Cysts develop in degenerative joint disease secondary to bone remodeling, and may occur as solitary (A) or multiple (B) lesions on either side of the joint. (Continued)
FIGURE 16.20. (Continued) Early myxoid tissue forms (C), which eventually cavitates (D). Microscopic features are nonspecific. Bland, sparsely cellular membranes with either loose or dense connective tissue walls are characteristic (E).
TABLE 16.6 Scoring Systems Used for the Histopathologic Assessment of Degenerative Joint Disease
I. Surface
A. Normal
B. Roughened, surface irregularities
C. Fibrillated, fissured
D. Clefts, erosions
1. Clefts to transitional zone
2. Clefts to radial zone
3. Clefts to calcified zone
E. Full-thickness erosions
F. Complete loss or cartilage
II. Cellularity
A. Normal
B. Decreased 25%
C. Decreased 50%
D. No cells
III. Cell cloning
A. Normal
B. Several doublets
C. Doublets and triplets
D. Multiple cell rests
A. Normal
B. 1-5 cells/low-power field (×10)
C. 6-15 cells/low-power field (×10)
D. 16-25 cells/low-power field (×10)
E. >25 cells/low-power field (×10)
IV. Territorial alcianophilia (e.g., safranin O staining)
A. Normal
B. Increased or 25% decreased staining
C. Decreased 50%
D. No staining
V. Interterritorial alcianophilia
A. Normal
B. Increased or decreased staining
C. Decreased 50%
D. No staining
VI. Tidemark
A. Normal
B. Duplication of tidemark
C. Crossed by blood vessels
VII. Bone
A. Normal
B. Subchondral sclerosis
C. Cysts
D. Osteophytes
VIII. Osteophytes
A. None
B. Early cartilage metaplasia
C. Small exophytic bulge
D. Moderate-sized osteophyte
E. Large osteophyte
IX. Synovial membrane
A. Decrease in lining cells
B. Normal
C. Slight increase
>6 layers
>6 layers, some inflammation
>6 layers, marked inflammation
Adapted and modified from Armstrong SJ, Read RA, Ghosh P, et al. Moderate exercise exacerbates the osteoarthritic lesions produced in cartilage by meniscectomy: a morphologic study. Osteoarthr Cartil. 1993;1:89-96; Hannan N, Ghosh P, Bellenger C, et al. Systemic administration of glycosaminoglycan polysulphate (Arteparon) provides partial protection of articular cartilage from damage produced by meniscectomy in the canine. J Orthop Res. 1987;5:47-59; Mankin HJ, Dorman H, Lippiello L, et al. Biochemical and metabolic abnormalities in articular cartilage from osteo-arthritic human hips. II. Correlation of morphology with biochemical and metabolic data. J Bone Joint Surg Am. 1971;53:523-537; Colombo C, Butler M, O’Byrne E, et al. A new model of osteoarthritis in rabbits. I. Development of knee joint pathology following lateral meniscectomy and section of the fibular collateral and sesamoid ligaments. Arthritis Rheum. 1983;26:875-886; and Williams JM, Ongchi DR, Thonar E. Repair of articular cartilage injury following intraarticular chymopapain-induced matrix proteoglycan loss. J Orthop Res. 1993;11:705-716.)
FIGURE 16.21. Resurfacing of the femoral head [(A) gross] is possible by subchondrally derived fibrocartilaginous tufts (B). Growth is usually by endochondral ossification (C), and resurfacing can be substantial (D).
FIGURE 16.22. (A) Composite. (Continued)
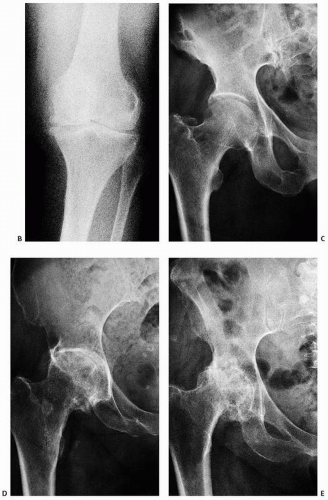
FIGURE 16.22. (Continued) (B) Roentgenographic features of degenerative joint disease. Characteristic findings are narrowing of the joint space, subchondral sclerosis, and cyst formation. (C-E) Varying degrees of degenerative joint disease, from mild to severe, the latter with abundant osteophyte formation (hip).
Intriguing etiologic candidates for the development of DJD include the reputed FAI. It is thought that FAI results from abnormal repetitive contact between the femoral head-neck junction and the acetabular rim as a result of abnormal bone morphology or activities requiring excessive hip flexion or internal rotation. “Cam” (femoral head-neck junction deformities), “Pincer” acetabular coverage abnormalities, or combined types have been postulated (33).
The natural history of OA is variable. The disease may stabilize or progress. Although joint replacement has been a major advance in bypassing crippling arthritis at a severely involved joint, no current satisfactory treatment is known to alter the course of the disorder.
Since articular cartilage has a poor intrinsic capacity to heal and renew itself, a plethora of surgical techniques and procedures have evolved to attempt to regenerate hyaline cartilage (34). Attempts to repair defects in articular cartilage have led to the use of cultured autologous chondrocytes. Alternatively, techniques such as microfracturing of the subchondral bone to stimulate bone marrow progenitor cells have been developed. To date, the challenge remains creating hyaline (not fibro-) cartilage. Reconstructing the three-dimensional integrity of normal cartilage is elusive as ever. Nonetheless, biological augments with cellular concentrates or platelet-rich plasma continue to be tried.
The treatment of OA is currently directed at eliminating pain and improving function. Recent evidence that OA and osteoporosis may represent different ends of a spectrum of metabolic bone pathology (35) underscores how delicate is the balance of nature.
There is compelling clinical evidence that several forms of OA are inherited. The following three types of OA are the most convincing:
Primary generalized OA characterized by Heberden and Bouchard nodes, premature degeneration of multiple joints, and symmetric, concentric, and uniform loss of articular cartilage at the knee and hip.
OA associated with familial cases of calcium pyrophosphate crystal deposition (chondrocalcinosis).
Stickler syndrome, a hereditary progressive arthro-ophthalmopathy.
Recent analyses by gene expression profiling hold promise that genetic links may be found (Fig. 16.23) (36). Likely candidate genes associated with osteoarthritis (OS) include collagens 1, 2, 9, and 11, cartilage oligomeric protein, aggrecan and various tissue growth factors (IGF-1, TGF-β1, IL-1R, IL-4R), and the secreted frizzled-related protein.
Variants of OA include cases with more inflammation and/or roentgenographic destruction. Both a rapidly destructive process (rapidly destructive DJD or “analgesic joint”) (Fig 16.24) and an erosive OA have been described.
The concept of a rapidly destructive arthrosis of the hip joint was first proposed by Postel and Kerboull in 1970. Most common as a unilateral disorder in elderly females, symptoms average about 1.4 years with joint space loss of 2 mm or more (or 50 percent of the joint space) within 1 year. Complete loss of the joint space averages 198 days (37). Radiographically, Postel disease may mimic a neuropathic or septic arthritis.
Only gold members can continue reading. Log In or Register to continue