Chapter 24 Arterial hypertension, angina pectoris, myocardial infarction and heart failure
• Hypertension and angina pectoris: how drugs act.
• Drugs used in both hypertension and angina.







• Acute coronary syndromes and myocardial infarction.
• Sexual function and cardiovascular drugs.
Hypertension: how drugs act
Consider the following relationship:
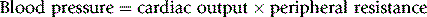
This being true, drugs can lower blood pressure by:
• Dilating arteriolar resistance vessels; achieved through direct relaxation of vascular smooth muscle cells, indirect relaxation by stimulating nitric oxide (NO) production, or by blocking the production or action of endogenous vasconstrictors, such as noradrenaline/norepinephrine and angiotensin.
• Dilating venous capacitance vessels; reduced venous return to the heart (preload) leads to reduced cardiac output, especially in the upright position.
• Reduction of cardiac contractility and heart rate.
• Depletion of body sodium. This reduces plasma volume (transiently), and reduces arteriolar response to noradrenaline/norepinephrine.
Angina pectoris: how drugs act
The supply of myocardial oxygen can be increased by:
• slowing the heart (coronary flow, uniquely, occurs in diastole, which lengthens as heart rate falls).
Drugs used in hypertension and angina
Vasodilators
Organic nitrates
Organic nitrates (and nitrite) were introduced into medicine in the 19th century.1 De-nitration in the smooth muscle cell releases nitric oxide (NO), which is the main physiological vasodilator, normally produced by endothelial cells. Nitrodilators (a generic term for drugs that release or mimic the action of NO) activate the soluble guanylate cyclase in vascular smooth muscle cells and cause an increase in intracellular cyclic guanosine monophosphate (GMP) concentrations. This is the second messenger which alters calcium fluxes in the cell, decreases stored calcium and induces relaxation. The result is a generalised dilatation of venules (capacitance vessels) and to a lesser extent of arterioles (resistance vessels), causing a fall of blood pressure that is postural at first; the larger coronary arteries especially dilate. Whereas some vasodilators can ‘steal’ blood away from atheromatous arteries, with their fixed stenoses, to other, healthier arteries, nitrates probably have the reverse effect as a result of their supplementing the endogenous NO. Atheroma is associated with impaired endothelial function, resulting in reduced release of NO and, possibly, its accelerated destruction by the oxidised low-density lipoprotein (LDL) in atheroma (see Ch. 26).
The nitrates are generally well absorbed across skin and the mucosal surface of the mouth or gut wall. Nitrates absorbed from the gut are subject to extensive first-pass metabolism in the liver, as shown by the substantially higher doses required by that route compared with sublingual application (and explains why swallowing a sublingual tablet of glyceryl trinitrate terminates its effect). They are first de-nitrated and then conjugated with glucuronic acid. The t½ periods vary (see below), but for glyceryl trinitrate (GTN) it is 1–4 min. The de-nitration of GTN is in fact genetically determined as the enzyme responsible, a mitochondrial alcohol dehydrogenase, ALDH2, is polymorphic and in subjects carrying a common coding variant (E504K) sublingual GTN has reduced efficacy.2
to the characteristic vasodilator headache comes and goes quickly (hours).3 Ensuring that a continuous steady-state plasma concentration is avoided prevents tolerance. This is easy with occasional use of GTN, but with nitrates having longer t½ (see below) and sustained-release formulations it is necessary to plan the dosing to allow a low plasma concentration for 4–8 h, e.g. overnight; alternatively, transdermal patches may be removed for a few hours if tolerance is suspected.
An important footnote to the use of nitrates (and NO dilators generally) has been the marked potentiation of their vasodilator effects observed in patients taking phosphodiesterase (PDE) inhibitors, such as sildenafil (Viagra) and tadalafil (Cialis). These agents target an isoform of PDE (PDE-5) expressed in the blood vessel wall. Other methylaxanthine PDE inhibitors, such as theophylline, do not cause a similar interaction because they are rather weak inhibitors of PDE-5, even at the doses effective in asthma. A number of pericoital deaths reported in patients taking sildenafil have been attributed to the substantial fall in blood pressure that occurs when used with a nitrate. This is an ironic twist for an agent in first-line use in erectile dysfunction that was originally developed as a drug to treat angina.4
Glyceryl trinitrate (see also above)
Calcium channel blockers
Calcium is involved in the initiation of smooth muscle and cardiac cell contraction, and in the propagation of the cardiac impulse. Actions on cardiac pacemaker cells and conducting tissue are described in Chapter 25.
Contraction of these cells requires an influx of calcium across the cell membrane. This occurs through voltage-operated ion channels (VOCs) and this influx is able to trigger further release of calcium from intracellular stores in the sarcoplasmic reticulum. The VOCs have relatively long opening times and carry large fluxes; hence they are usually referred to as L-type channels.6 The rise in intracellular free calcium results in activation of the contractile proteins, myosin and actin, with shortening of the myofibril and contraction of smooth muscle. During relaxation calcium is released from the myofibril and either pumped back into the sarcoplasm or lost through Na/Ca exchange at the cell surface.
There are three structurally distinct classes of calcium channel blocker:
Indications for use
• Hypertension: amlodipine, isradipine, nicardipine, nifedipine, verapamil.
• Angina: amlodipine, diltiazem, nicardipine, nifedipine, verapamil.
• Cardiac arrhythmia: verapamil.
• Raynaud’s disease: nifedipine.
• Prevention of ischaemic neurological damage following subarachnoid haemorrhage: nimodipine.
There has been some concern that the shorter-acting calcium channel blockers may adversely affect the risk of myocardial infarction and cardiac death. The evidence is based on case–control studies which cannot escape the possibility that sicker patients, i.e. with worse hypertension or angina, received calcium channel blockade. The safety and efficacy of the class has been strengthened by the recent findings of two prospective comparisons with other antihypertensives.7
Individual calcium blockers
(t½ 2 h) is the prototype dihydropyridine. It selectively dilates arteries with little effect on veins; its negative myocardial inotropic and chronotropic effects are much less than those of verapamil. There are sustained-release formulations of nifedipine that permit once-daily dosing, minimising peaks and troughs in plasma concentration so that adverse effects due to rapid fluctuation of concentrations are lessened. Various methods have been used to prolong, and smooth, drug delivery, and bio-equivalence between these formulations cannot be assumed; prescribers should specify the brand to be dispensed. The adverse effects of calcium blockers with a short duration of action may include the hazards of activating the sympathetic system each time a dose is taken. The dose range for nifedipine is 30–90 mg daily. In addition to the adverse effects listed above, gum hypertrophy may occur. Nifedipine can be taken ‘sublingually’, by biting a capsule and squeezing the contents under the tongue. In point of fact, absorption is still largely from the stomach after this manoeuvre, and it should not be used in a hypertensive emergency because the blood pressure reduction is unpredictable and sometimes large enough to cause cerebral ischaemia (see p. 417).
has a t½ (40 h) sufficient to permit the same benefits as the longest-acting formulations of nifedipine without requiring a special formulation. Its slow association with L-channels and long duration of action render it unsuitable for emergency reduction of blood pressure where frequent dose adjustment is needed. On the other hand, an occasional missed dose is of little consequence. Amlodipine differs from all other dihydropyridines listed in this chapter in being safe to use in patients with cardiac failure (the PRAISE study).8
Angiotensin-converting enzyme (ACE) inhibitors, angiotensin (AT) II receptor blockers (ARBs) and renin inhibitors
Uses
(see p. 406). ACE inhibitors have a useful vasodilator and diuretic-sparing (but not diuretic-substitute) action that is critical to the treatment of all grades of heart failure. Mortality reduction here may result from their being the only vasodilator that does not reflexly activate the sympathetic system.
The ARBs are at least as effective as ACE inhibitors in patients with heart failure and they can be substituted if patients are intolerant of an ACE inhibitor. Based on the Candesartan in Heart Failure Assessment of Reduction in Mortality and Morbidity (CHARM) trial, they may also benefit patients with heart failure and a low ejection fraction when added to treatment with a β-blocker and ACE inhibitor.9
In patients with type I (insulin-dependent) diabetes, hypertension often accompanies the diagnosis of frank nephropathy, and aggressive blood pressure control is essential to slow the otherwise inexorable decline in renal function that follows. ACE inhibitors appear to have a specific renoprotective effect, probably because of the role of angiotensin II in driving the underlying glomerular hyperfiltration.10 These drugs are now first-line treatment for hypertensive type I diabetics, although most patients will need a second or third agent to reach the rigorous blood pressure targets for this condition (see below). Their role in preventing the progression of the earliest manifestation of renal damage, microalbuminuria, is more complicated. Here the evidence suggests that ACE inhibitors do not slow the incidence of microalbuminuria in type I diabetics and an ARB may actually substantially increase it.10 In contrast, an ACE inhibitor halves the incidence of microalbuminuria in type 2 diabetics with hypertension and normal renal function on follow-up. A parallel group on verapamil did not show any protection confirming that inhibition of the renin–angiotensin–aldosterone (RAAS) axis is required for this effect, not simply lowering the blood pressure.11 For hypertensive type 2 diabetics with established nephropathy, both ARBs and ACE inhibitors protect against a decline in renal function and reduce macroproteinuria.10 The evidence suggests they are interchangeable in this respect. Whether combining the two classes of drugs (‘dual block’) confers further protection of renal function is not yet resolved, although ‘dual block’ does produce substantially better urine protein sparing than either agent alone.10
Following a myocardial infarction, the left ventricle may fail acutely from the loss of functional tissue or in the long term from a process of ‘remodelling’ due to thinning and enlargement of the scarred ventricular wall (see p. 425). Angiotensin II plays a key role in both of these processes and an ACE inhibitor given after MI markedly reduces the incidence of heart failure. The effect is seen even in patients without overt signs of cardiac failure, but who have low left ventricular ejection fractions (< 40%) during the convalescent phase (3–10 days) following the MI. Such patients receiving captopril in the SAVE trial,12 had a 37% reduction in progressive heart failure over the 60-month follow-up period compared with placebo. The benefits of ACE inhibition after MI are additional to those conferred by thrombolysis, aspirin and β-blockers. ARBs also prevent remodelling and heart failure in post-MI patients, but there is no additional benefit from ‘dual blockade’.13
Cautions
Certain constraints apply to the use of ACE inhibitors:
• Heart failure: severe hypotension may result in patients taking diuretics, or who are hypovolaemic, hyponatraemic, elderly, have renal impairment or with systolic blood pressure of less than 100 mmHg. A test dose of captopril 6.25 mg by mouth may be given because its effect lasts for only 4–6 h. If tolerated, the preferred long-acting ACE inhibitor may then be initiated in low dose.
• Renal artery stenosis (RAS, whether unilateral, bilateral renal or suspected from the presence of generalised atherosclerosis): an ACE inhibitor may cause renal failure and is contraindicated. ARBs are not necessarily any safer in this situation, because angiotensin II-mediated constriction of the efferent arteriole is thought to be crucial to the maintenance of glomerular perfusion in RAS.
• Aortic stenosis/left ventricular outflow tract obstruction: an ACE inhibitor may cause severe, sudden hypotension and, depending on severity, is relatively or absolutely contraindicated.
• Pregnancy represents an absolute contraindication (see below).
Adverse effects
• Persistent dry cough occurs in 10–15% of patients.
• Urticaria and angioedema (less than 1 in 100 patients) are much rarer, occurring usually in the first weeks of treatment. The angioedema varies from mild swelling of the tongue to life-threatening tracheal obstruction, when subcutaneous adrenaline/epinephrine should be given. The basis of the reaction is probably pharmacological rather than allergic, due to reduced breakdown of bradykinin.
• Impaired renal function may result from reduced glomerular filling pressure, systemic hypotension or glomerulonephritis, and plasma creatinine levels should be checked before and during treatment.
• Hyponatraemia may develop, especially where a diuretic is also given; clinically significant hyperkalaemia (see effect on aldosterone above) is confined to patients with impaired renal function.
• ACE inhibitors cause major malformations in the first trimester and are fetotoxic in the second trimester, causing reduced renal perfusion, hypotension, oligohydramnios and fetal death (see Pregnancy hypertension, p. 417).
• Neutropenia and other blood dyscrasias occur. Other reported reactions include rashes, taste disturbance (dysguesia), musculoskeletal pain, proteinuria, liver injury and pancreatitis.
Individual drugs
include cilazapril, fosinopril, imidapril, lisinopril, moexipril, perindopril, quinapril, ramipril and trandolapril. Of these, lisinopril has a marginally longer t½ than enalapril (it is the lysine analogue of enalaprilat), probably justifying its popularity as a once-daily ACE inhibitor. Some of the others are longer acting, with quinapril and ramipril also having a higher degree of binding to ACE in vascular tissue. The clinical significance of these differences is disputed. In the Heart Outcomes Prevention Evaluation (HOPE) study of 9297 patients, ramipril reduced, by 20–30%, the rates of death, myocardial infarction and stroke in a broad range of high-risk patients who were not known to have a low ejection fraction or heart failure.14 The authors considered (probably erroneously) that the results could not be explained entirely by blood pressure reduction.
in clinical use include candesartan, eprosartan, irbesartan, telmisartan, valsartan and olmesartan. Some of these may be marginally more effective than losartan at lowering blood pressure, but few if any comparisons have been performed at maximal dose of each drug. Losartan is generally used in combination with hydrochlorothiazide. In a landmark study this combination was 25% more effective than atenolol plus hydrochlorothiazide in preventing stroke.15
The cautions listed for the use of ACE inhibitors (above) apply also to AT1-receptor blockers.
Individual drugs
is the only orally active non-peptide renin inhibitor licensed (t½ 40 h). The agent is well tolerated apart from dose-dependent diarrhoea; it is not clear if this is a class side-effect. It produces additive effects on blood pressure with ACE inhibitors, ARBs, calcium channel blockers and thiazide diuretics. There are currently no outcome data in terms of preventing hypertension-related cardiovascular events, so it should be reserved for inhibiting the RAAS where an ACE inhibitor or ARB is not tolerated.16
Other vasodilators
Sodium nitroprusside is used in hypertensive emergencies, refractory heart failure and for controlled hypotension in surgery. An infusion17 may begin at 0.3–1.0 micrograms/kg/min, and control of blood pressure is likely to be established at 0.5–6.0 micrograms/kg/min; close monitoring of blood pressure is mandatory, usually by direct arterial monitoring; rate changes of infusion may be made every 5–10 min.
is effective through two actions: it acts as a nitrate by activating cyclic GMP (see above) but also opens the ATP-dependent potassium channel to allow potassium efflux and hyperpolarisation of the membrane, which reduces calcium ion entry and induces muscular relaxation. It is indicated for use in angina, where it has similar efficacy to β-blockade, nitrates or calcium channel blockade. It is administered orally and is an alternative to nitrates when tolerance is a problem, or to the other classes when these are contraindicated by asthma or cardiac failure. Adverse effects to nicorandil are similar to those of nitrates, with headache reported in 35% of patients. It is the only antianginal drug for which at least one trial has demonstrated a beneficial influence on outcome.18
is an alkaloid present in opium, but is structurally unrelated to morphine. It inhibits phosphodiesterase and its principal action is to relax smooth muscle throughout the body, especially in the vascular system. It is occasionally injected into an area where local vasodilatation is desired, especially into and around arteries and veins to relieve spasm during vascular surgery and when setting up intravenous infusions. It is also used to treat male erectile dysfunction (see p. 465).
Vasodilators in peripheral vascular disease
Night cramps occur in the disease and quinine has a somewhat controversial reputation in their prevention. Nevertheless, meta-analysis of six double-blind trials of nocturnal cramps (not necessarily associated with peripheral vascular disease) shows that the number, but not severity or duration of episodes, is reduced by a night-time dose.19 The benefit may not be seen for 4 weeks.
Adrenoceptor-blocking drugs
Adrenoceptor-blocking drugs occupy the adrenoceptor in competition with adrenaline/epinephrine and noradrenaline/norepinephrine (and other sympathomimetic amines) whether released from stores in nerve terminals or injected. There are two principal classes of adrenoceptor, α and β: for details of receptor effects see Table 23.1.
α-Adrenoceptor-blocking drugs
There are two main subtypes of α adrenoceptor:
• ‘Classic’ α1 adrenoceptors, on the effector organ (post-synaptic), mediate vasoconstriction.
• α2 Adrenoceptors are present both on some effector tissues (post-synaptic) and on the nerve ending (pre-synaptic). The pre-synaptic receptors (or autoreceptors) inhibit release of chemotransmitter (noradrenaline/norepinephrine), i.e. they provide negative feedback control of transmitter release. They are also present in the CNS.
For use in prostatic hypertrophy, see page 619.


The converse of the benefit in the treatment of prostatism is the adverse effect of urinary incontinence in women. Other adverse effects of α-adrenoceptor blockade are postural hypotension, nasal stuffiness, red sclerae and, in the male, failure of ejaculation. They may also exacerbate symptoms of angina.20 Effects peculiar to each drug are mentioned below.
Notes on individual drugs
is an irreversible non-selective α-adrenoceptor-blocking drug whose effects may last for 2 days or longer. The daily dose must therefore be increased slowly. It is impossible to reverse the circulatory effects by secreting noradrenaline/norepinephrine or other sympathomimetic drugs because its effects are insurmountable. This makes it the preferred α-blocker for treating phaeochromocytoma (see p. 419).
Indigestion and nausea can occur with oral therapy, which is best given with food.
has both α- and β-receptor-blocking actions that are due to different isomers (see β-adrenoceptor block, below). Its parenteral preparation is valuable in the treatment of hypertension emergencies (see p. 416).
β-Adrenoceptor-blocking drugs
Actions
β-Adrenoceptor selectivity
Some β-adrenoceptor blockers have higher affinity for cardiac β1 receptors than for cardiac and peripheral β2 receptors (Table 24.1). The ratio of the amount of drug required to block the two receptor subtypes is a measure of the selectivity of the drug. (See note to Table 23.1, p. 384, regarding the use of the terms ‘β1 selective’ and ‘cardioselective’.) The question is whether the differences between selective and non-selective β-blockers confer clinical advantages. In theory β1-blockers are less likely to cause bronchoconstriction, but in practice few available β1-blockers are sufficiently selective to be safely recommended in asthma. Bisoprolol and nebivolol may be exceptions that can be tried at low doses in patients with mild asthma and a strong indication for β-blockade. There are unlikely ever to be satisfactory safety data to support such use. The main practical use of β1-selective blockade is in diabetics, where β2 receptors mediate both the symptoms of hypoglycaemia and the counter-regulatory metabolic responses that reverse the hypoglycaemia.
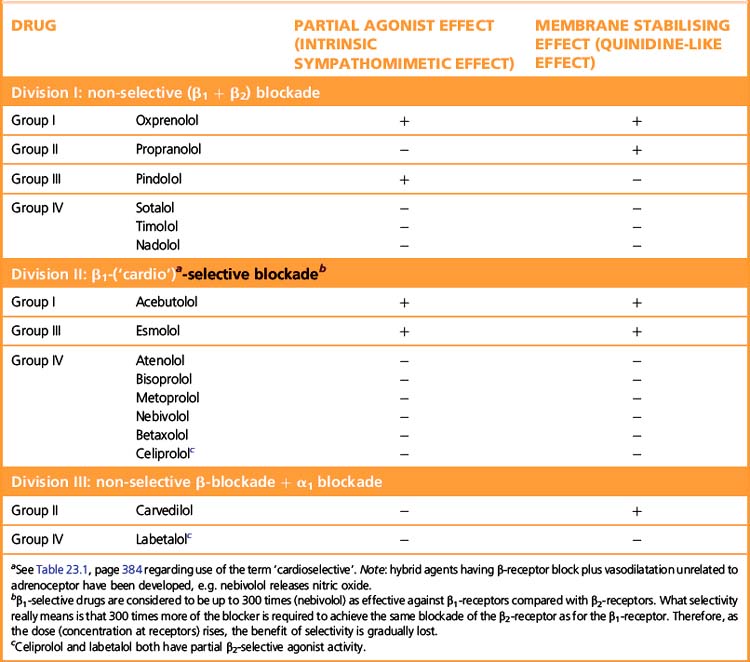
Some β-blockers (antagonists) also have agonist action or ISA, i.e. they are partial agonists. These agents cause less fall in resting heart rate than do the pure antagonists and may thus be less effective in severe angina pectoris where reduction of heart rate is particularly important. The fall in cardiac output may be less, and fewer patients may experience unpleasantly cold extremities. Intermittent claudication may be worsened by β-blockade whether or not there is partial agonist effect. Both classes of drug can precipitate heart failure, and indeed no important difference is to be expected because patients with heart failure already have high sympathetic drive (but note that β-blockade can be used to treat cardiac failure, p. 406).
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


