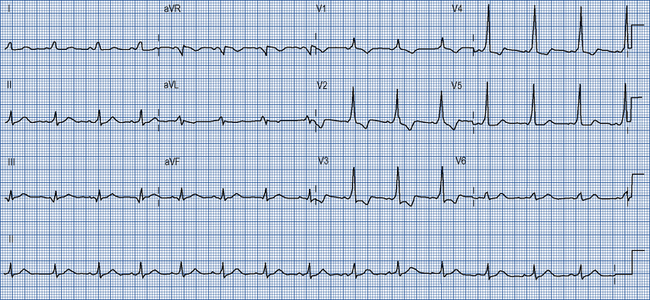
22 Arrhythmias
Normal cardiac electrophysiology
Cardiac action potential
The resting membrane potential of -60 to -90 mV occurs because the intracellular potassium (K+) concentration is much higher than the extracellular K+ concentration as a result of a transmembrane pump known as Na+–K+–ATPase, which pumps K+ ions into the cell in exchange for sodium (Na+) ions. K+ ions diffuse out of the cell through selective K+ channels (the inward rectifier current or IK1) unaccompanied by anions, resulting in a net loss of charge and thus a negative resting, diastolic or phase 4, transmembrane potential (Fig. 22.1A).
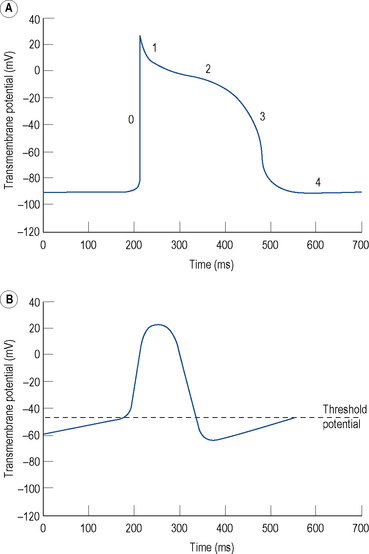
Certain specialised myocytes form the cardiac conduction system and these cells have pacemaker activity, that is, they are capable of generating their own action potentials due to gradual depolarisation of the transmembrane potential during diastole (phase 4), referred to as the pacemaker potential (Fig. 22.1B). The pacemaker potential occurs as a result of (i) a gradual reduction in an outward K+ current called the delayed rectifier (IK) current, (ii) increasing dominance of an inward current of Na+ and some Ca2+ ions known as If (f stands for ‘funny’) and (iii) an inward calcium current ICa through voltage-gated calcium channels. As a result of the pacemaker potential, the transmembrane potential gradually becomes less negative until a threshold potential is reached at which an action potential is triggered. The rate of depolarisation of the pacemaker potential, and hence the heart rate, is influenced by the autonomic nervous system. Sympathetic nervous system activation and circulating catecholamines increase the heart rate by binding to ß1-adrenoreceptors leading to an increase in intracellular cyclic AMP, which results in changes to the permeability of the various ion channels responsible for the pacemaker current. Parasympathetic nervous system activation, mediated by muscarinic cholinergic receptors, has the opposite effect.
The rapid depolarisation of the cardiac action potential (Fig. 22.1A, phase 0) occurs because of a rapid increase in the permeability of the cell membrane to Na+ ions, which enter rapidly through ‘fast’ Na+ channels in a current known as INa. The INa current is brief as the ‘fast’ Na+ channels inactivate rapidly. The early phase of repolarisation (phase 1) is due to closure of the fast Na+ channels, an outward K+ current known as Ito (to – transient outward) and a further K+ current known as the ultra-rapid component of the delayed rectifier current or IKur. The plateau phase (phase 2) of the cardiac action potential occurs because the inward movement of Ca2+ ions (ICa) is balanced by the outward movement of K+ ions. Repolarisation (phase 3) occurs as ICa diminishes and two further components of the delayed rectifier (IK) current known as the rapid (IKr) and slow (IKs) components predominate, with an important contribution from IK1.
There is considerable variation in the expression of trans-membrane ion channels in different parts of the heart, with corresponding variation in the morphology of the action potential. The most marked example is that myocytes in the sinus and AV nodes contain few Na+ channels. The upstroke of the action potential in these cells is due, predominantly, to the influx of Ca2+ ions and, therefore, is considerably slower than the upstroke in other myocytes (Fig. 22.1B). The variation in ion channel expression throughout the heart is essential for normal cardiac function, helps to explain the pathophysiology of many inherited and acquired diseases complicated by cardiac arrhythmia and accounts for the relative selectivity of antiarrhythmic and other drugs for certain parts of the heart.
Normal cardiac conduction
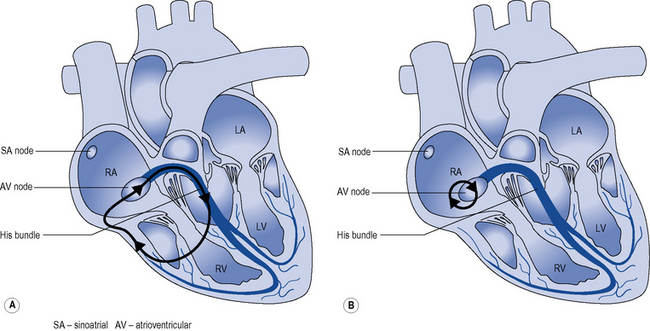
During normal sinus rhythm (Fig. 22.2), an activation wavefront begins in the sinus node, a group of cells with pacemaker activity on the upper free wall of the right atrium. The rate of diastolic depolarisation and hence the rate of discharge of the sinus node is increased by sympathetic nerve stimulation, circulating catecholamines or sympathomimetic drugs mediated by ß1-adrenoreceptors on the cell membranes of the sinus node myocytes. Parasympathetic (vagus) nerve stimulation exerts the opposite effect, mediated by muscarinic cholinergic receptors.
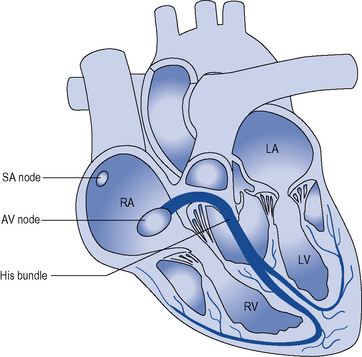
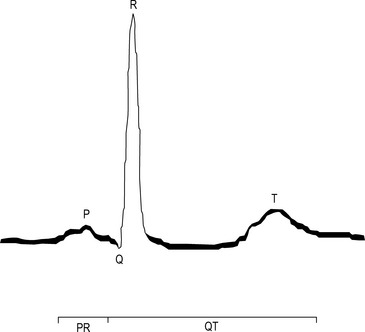
An activation wavefront spreads across the atrial myocardium, leading to atrial contraction and generating the P wave on the surface electrocardiogram (ECG; Fig. 22.3). The last part of the atria to be activated is the atrioventricular (AV) node, the electrical and structural properties of which result in a slow conduction velocity, allowing atrial emptying to be completed before ventricular contraction begins and represented by the PR interval on the ECG. Conduction velocity in the AV node is increased by sympathetic nerve stimulation, circulating catecholamines or sympathomimetic drugs, mediated by ß1-adrenoreceptors while parasympathetic (vagus) nerve stimulation exerts the opposite effect via muscarinic cholinergic receptors.
Arrhythmia mechanisms
Cardiac arrhythmias occur because of abnormalities of impulse formation or propagation.
Abnormal impulse formation
Abnormal automaticity
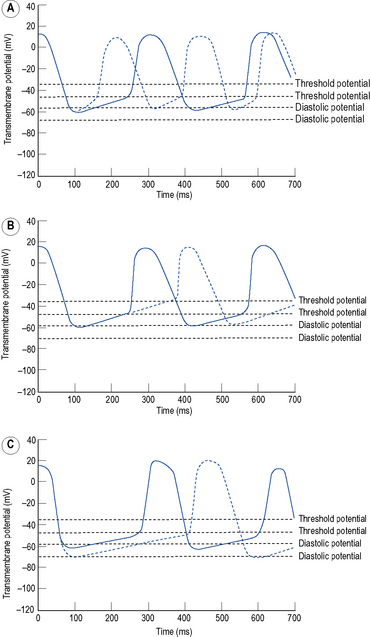
Automaticity is another term for pacemaker activity, a characteristic possessed by all cells of the specialised cardiac conduction system during health and, potentially, by other cardiac myocytes during certain disease states. The rate of firing of a pacemaker cell is largely determined by the duration of the phase 4 diastolic interval (Fig. 22.4). This in turn is determined by (i) the maximum diastolic potential following repolarisation of the preceding action potential, (ii) the slope of diastolic depolarisation due to pacemaker currents and (iii) the threshold potential for generation of a new action potential. In the healthy state, there is a hierarchy of firing rates within the specialised conduction system with the highest rate in the sinus node followed by the AV node and then the His–Purkinje system. The sinus node is, therefore, the dominant pacemaker and determines the heart rate, while the pacemaker activity in the distal conduction system is ‘overdriven’ by the sinus node. Abnormal automaticity describes either accelerated pacemaker activity in cells of the distal cardiac conduction system such that they escape from overdrive suppression by the sinus node, or the development of pacemaker activity in cells that do not form part of the cardiac conduction system.
Triggered activity
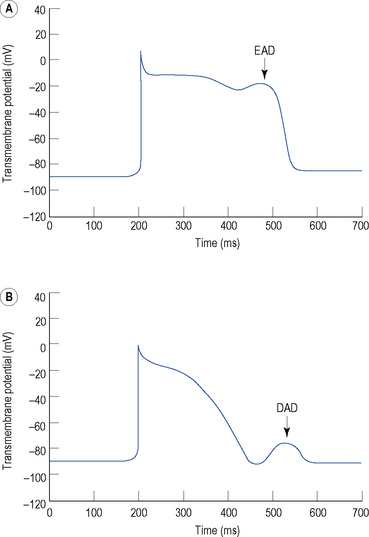
Triggered activity describes impulse formation dependent upon afterdepolarisations. Early afterdepolarisations (EADs) occur during phase 2 or 3 of the cardiac action potential whereas delayed afterdepolarisations (DADs) occur during phase 4 (Fig. 22.5). In both cases, afterdepolarisation may reach the threshold potential required for generation of a new action potential.
Abnormal impulse propagation
Re-entry
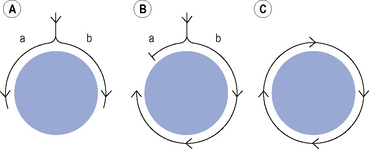
Many clinically important arrhythmias are due to re-entry, in which an activation wavefront rotates continuously around a circuit. Re-entry depends upon a trigger in the form of a premature beat, and a substrate, that is, the re-entry circuit itself. A precise set of electrophysiological conditions must be met in order for re-entry to occur (Fig. 22.6): (i) there must be a central non-conducting obstacle around which the re-entry circuit develops, (ii) a premature beat must encounter unidirectional conduction block in one limb (a) of the re-entry circuit, (iii) conduction must proceed slowly enough down the other limb (b) of the re-entry circuit that electrical excitability has returned in the original limb (a), allowing the activation wavefront to propagate in a retrograde direction along that limb, and (iv) the circulating activation wavefront must continue to encounter electrically excitable tissue. This is a function of the length of the re-entry circuit, the conduction velocity of the activation wavefront and the effective refractory period of the myocardium throughout the circuit. Class I antiarrhythmic drugs block sodium channels and, therefore, reduce the amplitude and rate of rise of the cardiac action potential and in so doing, reduce the conduction velocity of an activation wavefront. Class I antiarrhythmic drugs may exert their major antiarrhythmic effect by abolishing conduction altogether in areas of diseased myocardium forming part of a re-entry circuit in which conduction is already critically depressed. Class III antiarrhythmic drugs prolong cardiac APD and hence the refractory period. If previously activated cells in a re-entry circuit (the ‘tail’) remain refractory when the re-entrant wavefront (the ‘head’) returns to that area, conduction will fail and re-entry will be abolished. Drug-induced prolongation of the refractory period may, therefore, terminate and/or prevent re-entrant arrhythmias.
Clinical problems
Patients with a cardiac arrhythmia may present with a number of symptoms:
Diagnosis
Management
Supraventricular tachycardias
These are tachycardias arising from or involving the atria.
Atrial flutter
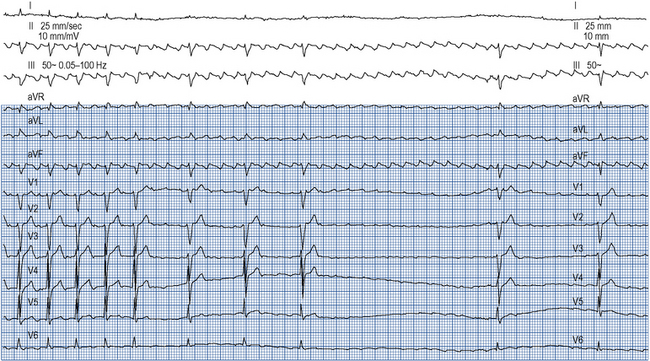
Atrial flutter confers a risk of thromboembolism similar to that of AF and this risk should be managed in the same way. Emergency management of atrial flutter is dictated by the clinical presentation but may include d.c. cardioversion or ventricular rate control with drugs which increase the refractory period of the AV node such as ß-blockers, verapamil, diltiazem or digoxin. ß-Blockers, verapamil and digoxin may be given intravenously. As the re-entry circuit is confined to the right atrium and does not involve the AV node, adenosine will not terminate atrial flutter but will produce transient AV block, allowing the characteristic flutter waves to be seen on the ECG (Fig. 22.7).
Junctional re-entry tachycardia
Two mechanisms account for most junctional re-entry tachycardias: both involve a macroreentry circuit (Fig. 22.8). AV nodal re-entry tachycardia (AVNRT) rotates around a circuit including the AV node itself and the so-called AV nodal fast and slow pathways, which feed into the AV node. Atrioventricular re-entry tachycardia (AVRT) comprises a re-entry circuit involving the atrial myocardium, the AV node, the ventricular myocardium and an accessory pathway, a congenital abnormality providing a second electrical connection between the atria and ventricles in addition to the His bundle, thus forming a potential re-entry circuit.
Many accessory pathways conduct only retrogradedly from the ventricles to the atrium. In these cases, the ECG during sinus rhythm appears normal and the accessory pathway is described as ‘concealed’. Other accessory pathways conduct anterogradely and retrogradely. In these cases, the ECG during sinus rhythm is abnormal and is described as having a Wolff–Parkinson–White pattern (Fig. 22.9). This abnormality is characterised by a short PR interval as the conduction velocity of an accessory pathway is usually faster than that of the AV node, and a delta wave, a slurred onset to the QRS complex which occurs because an accessory pathway inserts into ventricular myocardium which conducts more slowly than the His–Purkinje system.