Here, I is the maximum intensity of the depletion beam, and the saturation intensity I S is the intensity required to reduce the fluorescence probability by half [4]. Thus, larger depletion laser intensities will not only increase the likelihood of stimulated emission to occur but will also increase the resolution of the microscope by broadening the effective area of the depletion doughnut. Thus, STED microscopy allows the immediate acquisition of diffraction-unlimited images without requiring further computational post processing, as it is the case for other fluorescence nano-resolution techniques.
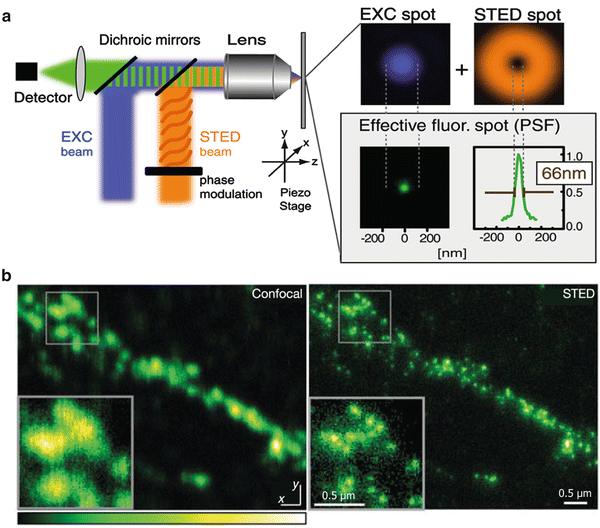
Fig. 1
STED microscopy breaks Abbe’s diffraction barrier. (a) Working principle of a STED microscope. The sample is illuminated with an excitation beam (blue ) and a doughnut-shaped depletion beam (orange ) that are spatially aligned. In the center of the depletion beam, spontaneous fluorescence is allowed to occur, whereas fluorophores in the bright surrounding area are depleted. The result is a lateral reduction of the effective excitation volume with concomitant improvement of resolution. (b) STED microscopy applied on biological samples. Neuronal synaptic terminals revealed by immunostaining of the synaptic vesicle protein synaptotagmin were imaged with confocal and STED microscopy for comparison. Individual terminals can be differentiated only under the increased resolution of STED microscopy, due to their subdiffraction size. Reproduced with permission from Ref. [7]
The principle of STED microscopy was first published by Stefan Hell and Jan Wichmann in 1994 [5]. In 2000 the technique was applied to living Saccharomyces cerevisiae and Escherichia coli cells labeled with organic dyes [3]. Since then, STED has been applied to diverse biological preparations, especially in the neuroscience field (see Fig. 1b). Once STED had successfully been applied on immunostained microtubules [6], later studies using immunofluorescence became relatively trivial. For example, the vesicle membrane protein synaptotagmin I was shown to remain clustered in the plasma membrane after vesicle fusion in cultured mammalian neurons [7]. Also, the doughnut-shaped structure of Bruchpilot, a protein important for accurate synapse assembly in the neuromuscular junction of Drosophila, was described with STED by using immunostaining [8]. Live imaging of antibody-labeled synaptic vesicles (using synaptotagmin I antibodies) was possible in 2008, featuring imaging speeds of ~28 frames per second [9]. The adaptation of STED microscopy for use with the green fluorescent protein (GFP) [10] opened further pathways for studying living preparations. Thus, STED imaging of fluorescent proteins has been used to reveal the axon arbors of serotonergic neurons in intact living organisms like Caenorhabditis elegans [11] and to study movements of dendritic spines in both brain slices [4, 12, 13] and, finally, the living mouse brain [14].
This general summary evidences how STED microscopy has been improved to fulfill the requirements of nowadays cell biology research. However, it is important to highlight that subdiffraction resolution will not only depend on the microscope setup or the strength of the depletion laser but also on the thickness of the sample, the fluorophore selection, and the sample preparation. In this chapter we will focus on the technical aspects needed for obtaining high-quality images and points relevant to the data interpretation.
2 Materials
2.1 STED Setups
In the last decade STED has undergone major technical improvements and enhancements. The achievable resolution in the lateral and axial dimensions can be changed by using different phase masks, which are used to modify the wavefront of the depletion beam to generate the necessary depletion patterns. The most commonly used is the vortex phase plate, which creates the well-known doughnut pattern for increasing the lateral resolution [15, 16]. Two-color STED has been performed by combining two pairs of excitation and depletion beams (one pair for each fluorophore) [17–19] or by selecting the fluorophores thus that the same depletion beam could be used for both fluorophores [20]. Three-color STED imaging was achieved (using only two different laser pairs) by separating the used dyes both spectrally and by their excited state lifetimes. This way, two spectrally similar dyes (KK114 and Atto 647 N) could be distinguished, even though they were excited and depleted with the same pair of lasers [21]. Such multicolor STED imaging enables nanoscale colocalization studies of macromolecules in biological systems. Moreover, an isotropic spatial resolution of ~30 nm has been demonstrated by applying STED through two opposing high numerical aperture objectives thereby generating an almost spherical focal spot. This technique was coined isoSTED [20, 22].
STED has been used in conjunction with other microscopy techniques, thus proving to be a tool with versatile implementations in the biological field. Two-photon excitation has been combined with STED microscopy [23], becoming useful in the study of neuronal architecture in deep planes of brain slices [24]. STED and electron microscopy were used in a correlative microscopy study, where fluorescently tagged proteins first localized by STED microscopy were spatially overlaid with subcellular structures later revealed by electron microscopy, all in the same samples [25]. Furthermore, STED microscopy and fluorescence correlation spectroscopy (FCS) were combined to characterize the diffusion dynamics of single protein and lipid molecules on the plasma membrane of living cells [26].
STED microscopy has remained technically challenging, because most of the improvements and fine-tuning to the particular fluorophores and sample requirements required the construction of customized STED setups. In the meantime, however, Leica Microsystems (Wetzlar, Germany. www.leica-microsystems.com/products/super-resolution/) has been constantly developing and refining commercial STED microscopes, which are also offered as upgrade to their confocal counterparts.
2.1.1 STED with Pulsed Lasers
The first setup developed for STED microscopy uses synchronized trains of light pulses for excitation and depletion [3]. The optimal durations of such excitation and depletion pulses range in the order of tens to hundreds of picoseconds; see, for example, [27]. Typically, an excitation pulse is immediately followed by a depletion pulse in order to prevent early spontaneous fluorescence coming from fluorophores located outside of the doughnut center. Keeping the duration of the excitation-depletion cycle shorter than the excited state lifetime of the fluorophore reduces the probability of detecting those early spontaneous events, which is important for setups where photons are continuously gathered throughout the experiment. However, in cases where this situation cannot be prevented, photons can be selectively gathered right after the depletion pulse by using a time-gated detection system [28].
The temporal separation of excitation and depletion in a pulsed STED configuration implies that relatively low average power is required for depletion. As only a given pool of fluorophores is excited at very specific time points (by the pulsed excitation laser), the depletion beam is only needed at specific points in time, at a moderately high intensity. Although the STED beam intensity required for pulsed systems is two orders of magnitude larger than the one for single-photon excitation (i.e., conventional laser scanning confocal imaging), this is still three orders of magnitude lower than the one for multiphoton excitation [29], thus representing a moderate damage to biological probes. Another benefit of pulsed STED is that excited fluorophores can relax from triplet states between pulses before being re-excited. This diminishes fluorophore photobleaching (by reducing further excitation and damaging of dyes found in the triplet state) and thus allows repeated imaging rounds, important for time-lapse experiments.
Pulsed STED setups are fairly expensive due to the high cost of most pulsed laser sources. As a further drawback, many pulsed depletion lasers are typically tunable in the far-red spectral range (~700–1000 nm), limiting the readily usable fluorescent dyes to the red and infrared spectral range. Imaging more commonly used dyes in the bluer spectral range (e.g., GFP and yellow fluorescent protein YFP) requires additional efforts, such as using an optic parametric oscillator (OPO, for nonlinear frequency conversion) to attain shorter wavelength light (by doubling the frequency of the wavelength to make it match with the red-sided tail of emission of the fluorophore) [7, 10]. Alternative light sources that are easily tunable include stimulated Raman scattering fiber sources [30] or supercontinuum lasers [28, 31].
In most figures presented below, we used a Leica pulsed STED microscopy setup (Leica Microsystems GmbH, Mannheim, Germany), based on a TCS SP5 confocal microscope equipped with a 100× 1.4 NA HCX PL APO oil objective. This microscope uses a pulsed diode laser (18 mW, 80 MHz, 640 nm emission, PicoQuant, Germany) for excitation and a pulsed infrared titanium/sapphire (Ti:Sa) tunable laser (1 W, 80 MHz, 720–1000 nm) for depletion (Mai Tai Broadband, Spectra-Physics, Santa Clara, CA, USA). Detection devices include two ultrasensitive avalanche photodiodes and high-sensitivity, low-noise PMTs (see Subheading 2.2).
2.1.2 STED with Continuous Wave (CW) Lasers
With the aim of making STED more easily applicable, the technique has also been implemented with continuous wave lasers [32]. In this configuration, the spatially overlapped excitation and depletion beams are continuously illuminating the sample during image acquisition, making the setup much simpler to implement as no synchronization or modification of laser pulses is necessary. CW STED is more affordable and flexible, since continuous wave lasers are less expensive and are commonly available throughout the visual spectral range, broadening the palette of usable dyes.
The main disadvantage of CW STED is that since the excitation beam functions continuously, the population of excited fluorophores is continuously renewed and thus needs to be constantly “surrounded” by sufficient depletion photons—resulting in a depletion beam power 3–5 times larger than the time-averaged power of the pulsed STED laser [32]. This difficulty in depleting all excited fluorophores leads to a less-defined fluorescence contrast at the doughnut inner border, resulting in less sharp images [27, 33]. CW STED was first introduced using for depletion a Ti:sapphire laser operating in the CW mode at 750 nm [32], but later visible fiber lasers emitting at 592 nm were used instead [34]. Another difficulty of CW STED is that the constant illumination of the sample promotes bleaching of the dyes through dark state excitation, requiring dyes less prone to reach triplet dark states.
2.2 Detectors
Sensitive detectors are necessary for STED microscopy, as a smaller focal volume results in a smaller amount of photons emitted from the sampled spot. To compensate for the reduced signal, pixel dwell times can be increased and more sensitive detectors can be implemented, so as to record more photons from the same spot.
Most commonly, avalanche photodiodes (APDs) are used as detection devices in STED setups. However, hybrid detectors (e.g., GaAsP-based detectors) have been used more recently, since they combine the wide dynamic range of photomultipliers (PMTs) and the high sensitivity of APDs, producing more contrasted images [35].
Particularly bright samples can be imaged with PMTs, although resolution might be suboptimal due to lower signal to noise ratio. As mentioned above, gating systems can be included in the detection process, to avoid collection of early produced photons that could contaminate the image [33].
2.3 Fluorophores and Sample Labeling
As mentioned in the Introduction, STED microscopy could produce in theory an infinitely small excitation volume from which fluorophores could be detected, by arbitrarily increasing the depletion laser intensity. Nevertheless, the main limitation for the application of STED microscopy to biological preparations is the availability of dyes that are stable enough under increasing powers of the depletion laser. Moreover, suitable dyes should have a high quantum yield for good detection and should have appropriate fluorescence lifetimes, in case of pulsed STED. At the beginning of STED microscopy, an additional limitation was the matching of emission spectrum of the fluorophores with the available pulsing STED lasers. Dyes in the red side of the spectrum like RH-414 and Pyridine 4 were used for labeling the vacuolar membrane of yeast cells and E. coli membranes, respectively [3].
Atto dyes have been successfully used in the commercial Leica pulsed STED setup, with Atto 647 N and Atto 655 being the most widely used. In our hands, Atto 647 N has worked well in combination with Chromeo 494 for two-color Leica pulsed STED. Technical improvements to double the wavelength frequency of a Ti:Sa pulsing laser allowed depletion of Alexa Fluor 594 [24]. With the advent of STED with CW lasers, blue-shifted fluorophores like Alexa Fluor 488 could be also used [34]. Dyes recommended for the Leica CW setup include BD Horizon V500, Oregon Green 488, Chromeo 488, Chromeo 505, and Atto 488, among others (see Note 1 ).
Most of the aforementioned dyes have been used conjugated to antibodies for immunofluorescence assays of fixed or live samples, easing the study of intermolecular interactions and structural details at the subcellular level. A big step in the study of live cells not only in culture but also in tissues was the application of STED on fluorescent proteins. The first report used a GFP-tagged protein imaged in rotavirus-like particles and GFP fused to an ER-targeting sequence in mammalian cells [10]. Morphological plasticity of neuronal dendrites was assessed in YFP-transgenic mice [4, 13]. A more stable and brighter version of YFP, citrine, was also targeted to the ER and imaged in living cells to detect morphological changes of this organelle in time [36]. A new monomeric far-red fluorescent protein, TagRFP657, was engineered by site-specific and random mutagenesis to fit into the excitation and depletion wavelengths of the commercial pulsing STED microscope [37]. The reversible switchable fluorescent proteins (RSFPs), also called photochromic fluorescent proteins, Dronpa and Padron were combined for double-label STED microscopy. Since both proteins are spectrally similar, only one excitation (488 nm) and one depletion beam (595 nm) were needed in combination with a beam at 405 nm that switches Padron off and Dronpa on [38].
Other mechanisms for protein labeling include genetically encoded tags. The SNAP-tag technology is based on the reaction of the protein O6-alkylguanine-DNA alkyltransferase for DNA repairing. The reaction can be used to transfer fluorophores from benzylguanines (BG) to the SNAP-tag, which can be fused with a protein of interest. Vimentin, MAP2, caveolin, and connexin-43 were SNAP-tagged and labeled with tetramethylrhodamine for STED imaging [39]. Another type of tag is the fluorogen-activating proteins (FAPs), which render nonfluorescent organic dyes fluorescent upon specific binding. Thus, actin molecules fused with FAPs were evidenced by activation of malachite green added to the media of living HeLa cells [40].
3 Methods
3.1 Samples
Different types of biological samples, fixed or living, can be studied under the STED microscope (see Subheading 1). Thin samples prepared on glass coverslips, including cell monolayer cultures, organelles, or membrane sheets, are very easy to image. The close proximity of the objects to the glass excludes major aberrations due to changes in the refractive index along the sample.
For live cell imaging the sensitivity of the detection device and the speed of movement can be limiting. Despite this, time-lapse recordings have been performed to follow morphological changes of the endoplasmic reticulum in living cells [36] or to track single vesicle fast movements in synaptic boutons and axons at video-rate imaging [9].
In thick preparations (e.g., tissue sections or in vivo imaging) the differences in refractive index between the oil-immersion medium and the biological specimen reduce the STED effect when focusing at deeper planes. Glycerol-immersion objectives with a correction collar have been successfully used to overcome this type of aberration, allowing subdiffraction resolution as deep as 120 μm into living brain slices [12] or up to 15 μm in the living mouse brain [14]. Nonetheless, these studies required abundant cytosolic expression of EYFP in neurons to get a sufficiently bright signal that could be imaged at such depths.
Immunostaining of thick fixed samples can be performed as for confocal microscopy, but special attention should be paid to the selection of the embedding medium (see below).
3.2 Embedding Procedures
Thin fixed samples (e.g., cell cultures, membrane sheets, isolated organelles) can be easily embedded in Mowiol, a polyvinyl alcohol-based, water-soluble polymer. For thicker samples the fluorescence intensity and STED resolution will drop at deeper planes due to spherical aberrations caused by a mismatch between the refractive index (n) of the embedding medium and the ones of the glass coverslip (n = 1.515) and the immersion oil (n = 1.518). Commonly used embedding media like Mowiol (n = 1.49) and glycerol (n = 1.45) are therefore not suitable [41]. 2,2′-Thiodiethanol (TDE) has been characterized as the best embedding medium to solve this problem, since its n can be easily adjusted by dilution with water (n = 1.515 for a 97 % TDE solution). TDE also compensates for changes of n generated by large cellular components and preserves the quantum yield of many fluorophores [41]. Drawbacks of TDE embedding are short life of samples (1–2 weeks) and higher mobility compared to Mowiol embedding (see Note 2 ). Figure 2 shows differences in STED resolution at different depths between a sample embedded in Mowiol and one embedded in TDE.

Fig. 2
TDE embedding preserves STED resolution along the axial plane in thick samples. The cranial muscle levator auris longus, responsible for ear movement, was dissected from a mouse. After immunostaining against the protein complexin (involved in the regulation of synaptic vesicle fusion), sections of the muscle were embedded in Mowiol (see Subheading 3.2.1) or TDE (see Subheading 3.2.2) for comparison. Complexin was imaged at the neuromuscular junctions (NMJs) by STED microscopy at four different planes: at the surface of the muscle and around 10, 20, and 30 μm of depth into the tissue. Note that STED resolution decreases at deeper planes of the Mowiol-embedded sample, due to spherical aberrations produced when the light beam reaches a medium of a refractive index different to those of the oil and the glass coverslip. In contrast, the perfect match in refractive index offered by TDE allows imaging at deep planes with preserved STED resolution
For even bigger fixed samples like whole brain preparations or small organisms, tissues can be embedded in a more stable matrix to allow subsequent slicing in ultrathin sections. This is an extremely important procedure, as (1) such samples are rarely easy to image in high-resolution microscopy, and correct embedding and sectioning procedures remove this difficulty; (2) ultrathin sectioning can be performed down to at least ~40 nm, thus offering a trivial (and cheap) method of obtaining super-resolution in the Z-axis.
Several plastic resins are available in the market with different polymerization temperatures, hardness, and pH values. Preliminary tests with different resins are advisable according to the type of sample and fluorophores used. For example, glycol methacrylate (GMA) was selected for imaging fluorescent proteins expressed in C. elegans, due to its good penetration and polymerization at pH 8, important for stability of the fluorophore [25]. In our laboratory we have successfully used melamine, a highly water-compatible resin that does not require previous dehydration of the tissue, ensuring its better preservation. Melamine embedding followed by thin sectioning has also been used with cell cultures in order to improve axial resolution for 3D reconstruction [42]. Figure 3 shows STED imaging of melamine sections from immunostained neurons. We have also used the acrylic resin LR White for samples labeled with chemical dyes, although dehydration steps are required, preserving membranes more poorly.
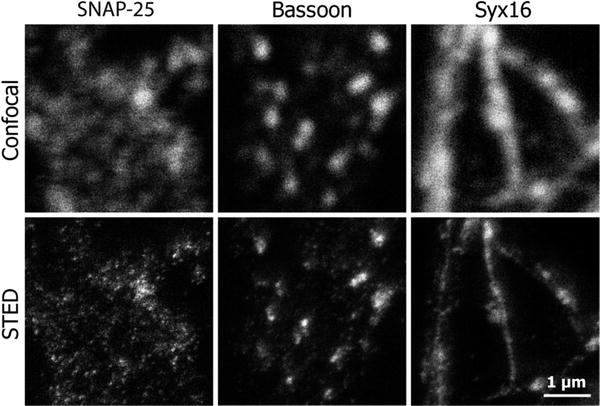
Fig. 3




Improving resolution in the Z-axis. Imaging large specimens (several millimeters thick) or generating high-resolution 3D reconstructions of cultured cells is possible using melamine embedding followed by ultrathin sectioning. This is an easy method to obtain subdiffraction axial resolution that complements the improved lateral resolution achieved with STED. As example, hippocampal neurons immunostained for the proteins synaptosomal-associated protein 25 (SNAP-25), bassoon, or syntaxin 16 (Syx16), embedded in melamine (see Subheading 3.2.3.) and sliced to 100–150 μm thick sections, were imaged in confocal and STED microscopy. The result in the STED image is clearly defined organelles, well differentiated from their surrounding environment, which is hardly achieved in the confocal image
Stay updated, free articles. Join our Telegram channel
Full access? Get Clinical Tree
