CHAPTER 13 Aplastic anemia and pure red cell aplasia
Acquired aplastic anemia
Acquired aplastic anemia (AA) is an uncommon disorder which in Europe and the United States affects about 2 per million of the population per annum; in other parts of the world including South-East Asia the incidence may be 2–3 times higher.1 Any age may be affected; there are two peaks, one occurring in adolescents and young adults, a second occurring after the age of 60, this bimodal distribution being most marked in white males.2,3,4 Male : female ratio is about equal but there may be a preponderance of males in the younger or adolescent age group.4
Definition and differential diagnosis
AA is defined by peripheral blood pancytopenia with a hypocellular bone marrow in which normal hemopoiesis is replaced to a greater or lesser extent by fat cells in the absence of genetic, malignant or predictable myelosuppressive causes. Remaining hemopoietic precursors and circulating blood cells are morphologically normal or show only minor abnormalities. The exclusions in the definition emphasize that other diseases may produce a similar morphological picture and these need to be excluded in coming to a diagnosis (Table 13.1). Hypoplastic myelodysplastic syndrome (MDS) may be particularly difficult to distinguish; the distinction may not be critical since there is considerable overlap in the pathogenesis and management of both conditions.
Table 13.1 Differential diagnosis of acquired aplastic anemia (AA)
| Pathophysiology | Examples | Differential features |
|---|---|---|
| Inherited AA | Fanconi anemia | Chromosome fragility Dysmorphism Family history |
| Dyskeratosis congenita | Nail/skin changes Leukoplakia X-linked, family history Short telomere syndromes | |
| Malignant AA | Hypoplastic MDS | Blood cell morphology Cytogenetics |
| Acute leukemia (presenting as AA) | Spontaneous remission followed by leukemic relapse | |
| Toxic AA | Irradiation Chemotherapy Benzene | History of exposure |
| Immune mediated | Autoimmune pancytopenia Large granular lymphocytosis Acute graft-versus-host disease | Multiple autoantibodies Immunophenotype Post-transplant |
Hairy cell leukemia (HCL) is occasionally misdiagnosed as AA. The bone marrow aspirate in HCL is often aparticulate and dilute, hairy cells may be scanty and the marrow trephine appears to show hypocellularity, but the reticulin is increased and the hemopoietic tissue is not replaced by fat cells but by the abundant cytoplasm of hairy cells. Confusion may be compounded by inappropriate treatment of HCL with anti-thymocyte globulin (ATG). ATG may lead to a temporary response in HCL.5 Immunophenotyping will detect the HCL.
Etiology
Drugs
The list of drugs which have been recorded as precipitating aplastic anemia is long,6 but mostly only single or a few cases have been reported for each drug and the evidence against many of the drugs is slim. Some of the more commonly implicated drugs7–11 are listed in Table 13.2. A difficulty in determining the role of drug exposure is the delay between exposure and the identification of marrow damage. Typically there is a delay of 2–3 months between marrow injury and the onset of pancytopenia. AA may develop only after prolonged or repeated exposure to the drug. The association can only be made if it is certain the patient was exposed to the drug and that the causation had been noted before.
Table 13.2 Drugs implicated in aplastic anemia
| Drug group | Examples | References |
|---|---|---|
| Antibiotics | Chloramphenicol Sulfonamides Sulfsalazine Co-trimoxazole | 6, 9 |
| Anti-inflammatory agents | Gold salts Indomethacin, sulindac Diclofenac | 6, 10, 11 |
| Thyrostatic drugs | Carbimazole Thiouracils | 7 |
| Anti-convulsants | Felbamate Carbamazepine Hydantoins | 8 |
| Anti-diabetic agents | Sulfonylureas Chlopropamide Tolbutamide | 6 |
| Anti-platelet drugs | Ticlopidine Clopidogrel | |
| Occupational/Domestic substances | Benzene Hexachlorocyclohexane (Lindane) | 12–14 14 |
Industrial/domestic chemicals
Benzene is myelotoxic.12–14 Exposure to sufficient levels leads inevitably to marrow damage but there seems to be wide variation in the dose required to induce toxicity between individuals There is good epidemiologic evidence that chronic exposure to benzene causes AA, often progressing to MDS and acute leukemia. Some chemicals which have been implicated as a cause of AA are shown in Table 13.2. DDT has been implicated but considering its previously very widespread use and the paucity of reported cases it seems that this compound has little or no hematologic toxicity and that reports may well be confounded by the solvents, including benzene, in the preparation of DDT for spraying.
Viruses
Hepatitis. Hepatitis is a precursor of aplastic anemia in about 5–10% of cases in the West, perhaps double that in the Far East.12 In the majority of cases no specific virus can be identified and the association is based on clinical grounds and the presence of abnormal liver function tests. The delay between the clinical hepatitis and the onset of pancytopenia is of the order of 6–12 weeks, a similar period to that between drug exposure and aplasia. There is some suggestion that chloramphenicol administration followed by hepatitis is particularly likely to be associated with aplastic anemia.15 Further evidence to support the association is the finding that in one series over a quarter of patients who underwent orthotopic liver transplant for fulminant liver failure following viral hepatitis developed aplastic anemia whereas patients transplanted for other reasons had no marrow failure.16 The prognosis in these patients relates to the severity of the marrow depression and not the supposed viral agent. The patients respond equally well to immunosuppressive therapy or stem cell transplantation as others with the same degree of marrow failure. Occasionally familial AA may be precipitated in siblings by hepatitis.17
Epstein–Barr virus. EBV infection is commonly accompanied by neutropenia or thrombocytopenia probably of an immune origin. Rarely there may be true marrow aplasia which behaves like other cases of acquired disease,18 though within this group there are some patients who develop pancytopenia with marrow aplasia in whom there is spontaneous recovery in 4–6 weeks.
Pathophysiology
Normal hemopoiesis takes place in the specialized environment of the bone marrow where pluripotent hemopoietic stem cells (HSC) give rise to lineage specific committed progenitors which produce the mature cells for the circulation. There is an intimate relationship between the HSC and the cells of the microenvironment of the marrow, including osteoblasts, which is epitomized in the concept of the stem cell niche. The quiescent stem cell is protected within the niche; mobilized stem cells are able to repopulate the niche even after extensive trafficking. It is in this environment that self-renewal can take place. Differentiation and maturation are controlled by specific cytokines and growth factors which act on specific lineages. Self-renewal occurs close to endosteal surfaces. Maturation increases towards the central arteriole (see Chapters 2–4).
The stem cell in aplastic anemia
There are qualitative and quantitative abnormalities of hemopoietic stem cells in AA.19–23 Short-term colony assays of committed progenitor cells – colony-forming units (CFU-C, CFU-E), burst-forming units (BFU-E) – are markedly reduced in AA and remain low even after recovery.19–21 Committed progenitors, identified by long-term culture initiating cells (LTCIC) are also reduced and have a poorer proliferative potential than normal stem cells, with poorer survival of colony forming cells in long-term bone marrow culture.
The microenvironment in aplastic anemia
Long-term cultures depend upon a viable and confluent stroma for proper growth. Cross-over experiments have shown that stroma grown from aplastic anemia marrow can support colony-forming cells from normal marrow but that colony forming cells (CD34+) from aplastic marrow will not grow on either normal or aplastic marrow suggesting that the stroma is not at fault in the pathogenesis of AA.19,20 It is not always possible to grow stroma from AA and there may be a degree of heterogeneity in the pathophysiology.
Hemopoietic growth factor production is normal in AA.24,25 Erythropoietin (Epo), thrombopoietin (Tpo) and granulocyte colony stimulating factor (G-CSF) levels are increased though the concentration of circulating G-CSF is low in both normals and patients with AA. These observations might explain why Epo is ineffective in the treatment of AA whereas G-CSF may have a part to play in direct stimulation of bone marrow stem cells. A possible role for thrombopoietin receptor agonists in promoting platelet production in AA has yet to be determined.
It is clear that the stem cell in aplastic anemia is damaged but the precise nature of that damage and the subsequent changes which may lead to the evolution of abnormal clones are not fully understood.26,27 The observation that there is shortening of the telomeres in the myeloid series in acquired aplastic anemia28,29 and that marked shortening of telomeres is a feature of dyskeratosis congenita and other inherited types of aplastic anemia30,31 has suggested that damage to telomeres or the mechanisms which control their regeneration are consequences in the pathogenesis of aplastic anemia26,27 which may account for the increased risk of MDS and acute leukemias in both inherited and acquired syndromes.32
Autoimmune basis of aplastic anemia
AA has been considered a probable autoimmune disorder since the introduction of anti-thymocyte globulin (ATG) for successful immunosuppressive treatment of the disease.33 The process is thought to be mediated by CD8+ T-cells. Oligoclones of expanded CD8+ CD28− cells, directed against as yet unidentified antigens, recognize and induce apoptosis in autologous myeloid cells.34 In some instances the T-cells clones have been found to disappear on recovery. There is also some evidence that Tregs, which control auto-reactive T-cells, are reduced in AA,35 which may result in the dysregulation of autoimmunity.
Hematology
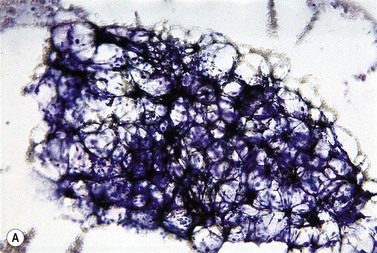
The peripheral blood film shows pancytopenia without gross morphological abnormalities in the remaining cells. There may be some macrocytosis of remaining red cells usually with an absolute reticulocytopenia. A relative reticulocytosis should always raise the possibility of associated paroxysmal nocturnal hemoglobinuria (PNH). Granulocytes often show increased staining of granules, the so-called toxic granulation of neutropenia. The neutrophil alkaline phosphatase score is increased (it falls if PNH develops). Monocytes may be reduced in proportion to the granulocytes. Platelets are reduced and of small and uniform size. There is usually a variable reduction in the lymphocyte count, but sometimes the count is normal or even increased so that the total white cell count may be normal. Abnormal cells are not seen. The bone marrow aspirate is normally easily obtained, typically with many fragments which have a lacy, empty appearance (Fig. 13.1A). The cell trails are hypocellular with a relative increase in lymphocytes and plasma cells and other non-hemopoietic forms. There may be a minor degree of dyserythropoiesis but in general remaining hemopoietic precursors are normal in appearance. Lymphocytes and plasma cells may appear to be increased but this is because of the lack of hemopoietic cells and there is no consistent increase in lymphocytes or in any subset in the marrow taken from patients with aplastic anemia. In the early stages of aplastic anemia, macrophages appear active with increased intracellular iron and erythrophagocytsis may be prominent. In a proportion of cases the hypocellularity of the marrow is patchy with areas of cellular marrow remaining. The bone marrow aspirate under these circumstances may be misleadingly cellular. A trephine biopsy (sometimes more than one) is necessary to assess cellularity properly.
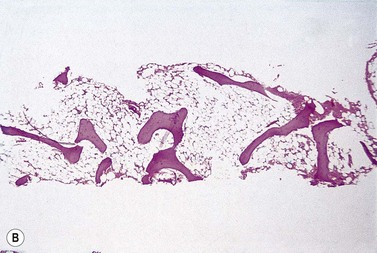
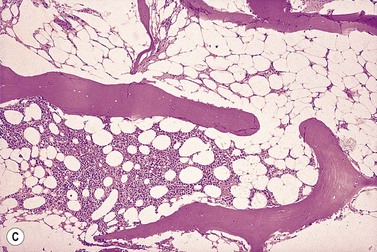
The trephine shows the fat replacement of marrow with or without the remaining islands of cellularity (Fig. 13.1B, C). The presence or absence of cellular ‘hot pockets’36 does not correlate with the severity of the peripheral blood pancytopenia and the apparent cellularity of a trephine specimen may not be reflected in the blood count. Non-hematopoietic cells remain, sometimes giving the impression of a chronic inflammatory infiltrate. Reticulin fibers are scanty, commensurate with the degree of hypocellularity. The architecture of the bone marrow remains essentially normal in distinction from the abnormal distribution of hemopoiesis in MDS.
Cytogenetic studies on bone marrow are required at presentation of patients with aplastic anemia, mainly to exclude hypoplastic MDS. Typically chromosome configuration and number are normal in aplastic anemia but it is now clear that in a number of cases there may be evidence for a clone of cytogenetically abnormal cells to be present at presentation which may be stable or transient but does not seem to affect response to treatment.37 However, in some series the development of trisomy 8 was associated with a good response to immunosuppression whereas monosomy 7 carried a poor prognosis with high probability of transforming to MDS or acute leukemia.38,39
Classification of aplastic anemia
A classification of the severity of the marrow damage in AA was devised in the 1970s to allow the comparison of the effectiveness of different treatments in this disease without a gross imbalance in the severity of the different groups.40 The original classification was based on observations of survival curves of collective series of drug-induced aplastic anemia which showed that there appeared to be two populations, one with a median survival of a few months and a 1-year survival of <10%, another which had a more prolonged survival, even though the patients remained with a degree of pancytopenia. Studies of these patients’ peripheral blood and bone marrow allowed prognostic features to be devised which identified the severe aplastic anemia (SAA) and the non-severe aplastic anemia (NSAA). It is clear that the degree of marrow damage is not a double population but a spectrum and improving support and treatments have led to modification of the classification into three groups, very severe aplastic anemia (VSAA), having been added41 (Table 13.3). The classification includes an assessment of the degree of hypocellularity of the marrow based on the trephine biopsy findings. This is the most subjective of the measurements, particularly as the cellularity may vary quite considerably in different samples and indeed in the same sample, the so called ‘hot pockets’.
Table 13.3 Gradation of severity of acquired aplastic anemia40,41
| Grade of severity | Definition | |
|---|---|---|
| Peripheral blood | *Bone marrow | |
| VSAA | Neutrophils <0.2 × 109/l Platelets < 20 × 109/l Reticulocytes <20 × 109/l Transfusion dependent | <25% normal cellularity. Moderately hypocellular <30%. Remaining cells hemopoietic. |
| SAA | Neutrophils <0.5 but >0.2 × 109/l Otherwise as for VSAA | As for VSAA |
| NSAA | Neutrophils <1.5 but >0.5 × 109/l Platelets <100 >20 × 109/l Reticulocytes <60 but >20 × 109/l | Hypocellular |
NSAA: non severe aplastic anemia; SAA: severe aplastic anemia; VSAA, very severe aplastic anemia.
* The bone marrow in aplastic anemia is often patchy in cellularity and the assessment of cellularity, even with a good trephine, may be difficult.
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


