Non-infectious a
Associated with vasculitides
With large-vessel vasculitis
Giant cell arteritisb
Takayasu arteritisb
With variable vessel vasculitis
Cogan’s syndromeb
Behçet’s diseasec
With medium- and small-vessel vasculitis
Polyarteritis nodosad
Wegener arteritisd
Microscopic polyangiitisd
Associated with systemic rheumatic disorders
HLA-B27 associated spondyloarthropathies
Ankylosing spondylitisb
Reiter syndromec
Replapsing polychondritisc
Systemic lupus erythematosusd
Rheumatoid arthritisd
Sarcoidosisd
“Single organ” vasculitis
Isolated idiopathic aortitis (thoracic)
Isolated idiopathic periaortitis (abdominal)
Idiopathic retroperitoneal fibrosis (Ormond disease)
Inflammatory abdominal aortic aneurysm
Radiation-induced aortitis
Infectious
Luetic (syphilis)
Mycobacterial (tubercolosis)
Bacterial
Salmonella spp.
Staphylococcus spp.
Streptococcus pneumonia
Infectious aortitis is a life-threatening disease, with high inherent risk of acute complications such as aortic aneurysm rupture; non-infectious forms however are often characterized by an indolent and insidious course, with progressive worsening, leading to significant quality-of-life limitations and potentially lethal evolutions. As a result of the different pathophysiological processes underlying the various forms of aortitis, it can in turn assume the phenotypes of dilative or obstructive disease of the aorta and its main branches, and present either isolated or within one of the several possible associated systemic syndromes, with or without involvement of other organs, eventually resulting in a myriad of diverse clinical pictures. A high index of diagnostic suspicion is necessary to avoid the complications and a correct differential diagnosis among the different etiologies is required to timely set the correct therapeutic strategy [1]. Diagnosis and differentiation can take advantage today of well-codified clinical criteria, at least for the most frequent forms of aortitis, multiple imaging modalities, laboratory tests and, to some extent, histology. In this chapter, we will address the description of aortitis and its treatment, emphasizing the above multiplicity of etiologies, involved mechanisms and clinical pictures, and focusing on the pharmacotherapy of the most common and notable forms of aortitis.
Non-infectious Aortitis
Non-infectious aortitis is more frequently encountered than infectious aortitis: inflammation is secondary to an autoimmune reaction in most of the non-infectious diseases, idiopathic in a minority of cases. The association between autoimmune (“rheumatic”) diseases and aortic involvement is well known, but the prevalence of aortic involvement in the different rheumatic diseases is quite variable: in some of them, namely large-vessel vasculitides, aortic involvement is, by definition, part of the canonical clinical picture (e.g. Takayasu arteritis, giant cell arteritis); in others (e.g. spondyloarthropathies or anti-neutrophil cytoplasmic antibody-related diseases) an arterial inflammation, possibly but not regularly involving also the aorta, can be observed as a part of a systemic or multiorgan involvement. In fact, according to the 2012 Chapel Hill Consensus Conference Nomenclature [2], vasculitides that affect large arteries more often than the others are named “large-vessel vasculitis”, those affecting predominantly medium caliber arteries are referred to as “medium-vessel vasculitis” and those affecting predominantly small size vessels are named “small-vessel vasculitis” (Table 2.1). Consistently, in 2006 a review on aortic involvement in rheumatic disorders listed Takayasu arteritis and giant cell arteritis among those most frequently affecting the aorta (>10 % patients), along with long-standing ankylosing spondylitis and Cogan syndrome; rheumatic diseases in which aortic involvement is an uncommon (<10 %) but well-documented complication include rheumatoid arthritis, seronegative spondyloarthropathies, Behçet disease and relapsing polychondritis; rheumatic diseases with isolated case reports of aortic involvement or uncertain involvement include sarcoidosis, antineutrophil cytoplasmic antibody-associated aortitis (Wegener granulomatosis and polyarteritis nodosa), and systemic lupus eryhematosus [3].
Giant-cell arteritis (GCA), also known as Horton’s (temporal) arteritis, is a chronic inflammatory large- and medium-vessel vasculitis that affects persons older than 50 years of age (reported male to female ratio ranges 1:2 to 2:3). GCA is much more common than Takayasu arteritis in the general population, with an estimated incidence of about 19 cases/million/year among patients over 50 years of age [4]. Although there is a markedly increased incidence of GCA in northern Europe and in populations with similar ethnic background [5], the disease can occur in all populations. The rate of aortic involvement in GCA is classically reported around 15–18 % (with a predominance of the ascending aorta, but possible involvement of the abdominal), however subclinical inflammation of the aorta may be present in a notably larger proportion of GCA patients. Branches most commonly involved are those arising from the external carotid artery, especially the superficial temporal artery, but ophthalmic, vertebral, coronary, and mesenteric arteries may also be involved.
The etiology of GCA is not well established. Polymorphisms of genes such as tumor necrosis factor-alpha (TNF-α), vascular endothelial growth factor (VEGF), endothelial nitric oxide synthase (eNOS), intercellular adhesion molecule (ICAM)-1, IL-6, and others appear to be more frequent in patients with GCA, although their pathogenetic role is still to be determined. It has been also postulated that a still unknown infectious pathogen may trigger the aberrant immune response [6].
The characteristic pathology feature of GCA is the presence of granulomatous inflammatory reaction in the vessel wall, with mostly macrophages, CD4+ T-cells and giant multinucleated cells constituting the granulomas, particularly located at the intima-media border, but also B-cells. Giant cells can actually be absent in 30–40 % cases. The CD4+ lymphocytes differentiate into T-helpers and produce interferon-gamma (IFN-γ), which activates macrophages, in turn producing reactive oxygen species and proteolytic enzymes, including matrix metalloproteases, the effectors of arterial wall elastic matrix degradation [7]. Lymphocytes and macrophages’ products, including TNF-α and IL-6, also enter the blood and are responsible for the systemic clinical syndrome in GCA (see next section). Alternatively or adjunctively, a systemic inflammatory response may stimulate pattern recognition receptors at vascular level thereby activating vascular dendritic cells, in turn initiating T-cell response. The intima may be thickened and the medial elastic laminae fragmented, whereas in the late stages intimal changes may be minimal and medial changes are largely constituted by fibrosis.
Also known as pulseless disease or Martorell syndrome, Takayasu arteritis (TKA) is a necrotizing and obliterative segmental, large-vessel panarteritis of unknown cause with a predilection for young women (>80 % of cases). Epidemiology varies in the different geographic areas: in the United States, incidence estimates from Olmstead County, Minnesota, are 2.6 cases/million/year, whereas in Sweden and Germany they are 1–1.2 cases/million/year [8] Autopsy studies in Japan document a much higher prevalence, with evidence of TKA in 1 every 3,000 individuals. Also, the age of disease onset differs: it is earlier (15–25 years) in Asians compared to European women (40 years) [9]. Involvement of the aorta is frequent, reported between 80 and >90 %. The most commonly affected aortic segment is the abdominal aorta, however ascending aorta involvement has been described as more typical in Japanese women [3].
Although the exact etiology of TKA is not well known, a prior tubercular or streptococcal infection, genetic factors, and autoimmune mechanisms (possible association with rheumatoid arthritis) have been implicated as etiological factors [9]. The pathogenetic mechanisms are unknown as well, although it is considered to be antigen-driven cell-mediated autoimmune processes, although the specific antigenic stimuli have not been identified [10]. Vessel injury occurs as a result of invasion by leukocytes (including T-lymphocytes, NK-cells, B-lymphocytes, macrophages and others) deriving from the vasa vasorum, migrating to the intimal layer and producing a number of cytokines, including interleukin-6 (IL-6), tumor necrosis factor-alpha (TNF-α), B-cell activating factor (BAFF) and others. Myointimal proliferation most commonly results, leading to stenosis of the vessel (most commonly the abdominal aorta) or its branches (especially supra-aortic vessels, iliac arteries and renal arteries), however medial smooth muscle cell necrosis and derangement of the extracellular matrix is another possible evolution, leading to aneurysm formation in 45 % cases (more common at the aortic root and ascending tract) [8]. Aside from the presence of granulomas, possibly including giant cells, in the aortic wall (particularly in the adventitia) and of perivascular cuffing of the vasa vasorum, histopathology usually reveals lymphoplasmacytic infiltrate of adventitia and media (Fig. 2.1), a non-specific finding present in a number of rheumatic disorders, such as relapsing polychondritis, systemic lupus erythematosus (SLE), ankylosing spondylitis, as well as in aortitis of infectious or toxic etiology. At late stages (at least >5 years of disease), unspecific wall calcification is observed, especially in the case of stenotic evolution.
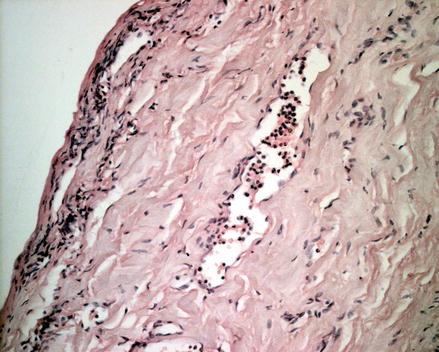
Figure 2.1
Histology findings of early stage non-infectious aortitis. Hematoxylin-eosin coloration shows multiple infiltrates of lymphocytes in the adventitia and sub-adventitial media, particularly surrounding the vasa vasorum, along with areas of initial focal medial elastic fibers disruption
Indeed, the pathology pictures of the different forms of aortitis show substantial overlap, and contribute to make differential diagnosis quite challenging. In this perspective, the stenotic lesions, with thickened intima and media, have been reported as pathognomonic of TKA [8].
Ankylosing Spondylitis (AS) is an HLA-B27 disease classified as a seronegative spondyloarthritis, since it is not characterized by circulating rheumatoid factor. Risk of aortic involvement (predominantly aortic valve, root, ascending aorta) increases with disease duration. Pathology studies of the sacroiliac joints in patients with AS have shown a prominent role of synovitis and subchondral myxoid bone marrow changes, processes mediated by activated T-lymphocyte-derived TNF-α and TGF-β, in initiating intra-articular joint destruction; a Klebsiella pneumoniae infection may participate in providing an antigen that reaches the synovia and initiates T-cell responses in genetically susceptible individuals [11]. Similar mechanisms could induce the typical fibrosis changes of AS-associated valvulitis and aortitis: in particular at the level of the proximal aorta, vasa vasorum narrowing, fibrotic changes in the adventitia, medial matrix disruption and fibrosis and myointimal proliferation ensue [12].
Cogan’s syndrome (CS) is a rare disease of young adults, with a mean age of 29 years at disease onset, defined by the presence of both ocular (keratitis) and inner ear inflammation [13]. Aortitis with valvulitis and aortic insufficiency has been documented as occurring from 2 weeks to 12 years after the initial diagnosis of the syndrome and has an estimated prevalence of up to 10 % [1, 3]. Etiopathogenesis is unknown: formerly believed to derive from a Clamydia spp. infection, today, after the finding of autoantibodies and lymphocyte activation against corneal and endothelial antigens, it is considered an autoimmune disorder [14]. Histologic analysis of the aortic wall reveals inflammation with prominent lymphocytic infiltration, destruction of medial elastic tissue, fibrosis, and neovascularization, which finally result in aneurysm formation [15].
Behçet disease (BD) is a rare, multisystemic, and chronic inflammatory disease of unknown etiology, characterized by mucocutaneous manifestations (aphtous ulcers), especially including genital and oral ulcers and often-severe sight-threatening inflammatory eye disease. It can be associated (up to 30–40 % cases) with vasculitic manifestations in arteries and veins of variable size [2]. The frequency of aortic involvement in BD varies according to different studies, ranging from 50 % of patients in reports from Turkey and Italy [16], to <1 % in other studies including only clinically significant aortic aneurysms [17]. Macroscopically, saccular aneurysms affecting the abdominal and/or thoracic aorta and their branches are typical of BD. Histopathology of the involved aorta shows lymphocytic infiltration mixed with histiocytes and eosinophils with giant cells around vasa vasorum of media and adventitia. Destruction of media leads to aneurysm formation and may proceed to pseudoaneurysm formation and rupture [18]. Aortitis derives not from direct large-vessel inflammation but rather due to vasculitis of the vasa vasorum that supply the vessel wall.
Apart from AS, other HLA-27-associated seronegative spondyloarthropathies can present with vascular involvement and potentially, but more rarely compared to the other abovementioned syndromes, with aortitis. These include Reiter’s syndrome, an arthritic disease of the lower limbs associated with typical cutaneous lesions and relapsing polychondritis, affecting proteoglycan rich tissues, such as cartilages and vessels. Less frequently, some of the anti-neutrophil cytoplasmic antibody- (ANCA-) related diseases, namely Wegener’s granulomatosis, microscopic polyangiitis and polyarteritis nodosa, which more typically involve small-size vessels, can be associated with large-vessel involvement and therefore aortitis. Whether aortitis in such cases is initiated by vasa vasorum involvement or by primitive inflammation of the intimal layer (“intimitis”) has not been clarified yet [19].
Following the 2012 Chapell Hill Consensus Conference Nomenclature [2], isolated idiopathic aortitis (more frequently observed at the abdominal level) should be classified within the group of “single organ” vasculitides. Idiopathic aortitis is characterized by aortic wall inflammation in absence of any systemic disease or infection, and it usually involves the ascending aorta and arch. This condition affects women more often than men (3:2 ratio) and is asymptomatic until it is discovered incidentally or by post-surgical histological analysis [20]. In those latter cases, macroscopic appearance can already suggest inflammatory etiology, i.e. by the typical diffuse irregular scarring of the intima, referred to as “tree-barking” sign [1]. Histology can vary, however overlapping with the spectrum of lesions already mentioned above for specific etiologies. Infiltrates can include macrophages, T-cells, B-cells and also giant multinuclear cells. Idiopathic inflammatory aneurysms of the abdominal aorta, constituting 5–25 % of all abdominal aneurysms [21], are characterized by thickening of the aortic wall, associated with a considerable peri-aortic reaction and dense adhesions. They are more frequently observed in young males with familiar history of aneurysm, and use of tobacco smoke. Retroperitoneal fibrosis is characterized by a chronic inflammation with fibrous tissue deposition in the retroperitoneum surrounding the aorta, the stem of its main abdominal branches and the ureters. Complications include hydronephrosis, aortic-enteric fistula, and secondary bacterial infections [1].
It has been recently discovered that some cases of idiopathic aortitis, idiopathic aneurysm and retroperitoneal fibrosis are actually secondary to a so-called “IgG4-related systemic disease”, in which multiple organs are involved by inflammatory infiltrates constituted by IgG4-expressing plasmacells, and abnormally high levels of IgG4 are found in the serum. This syndrome was first described in patients with autoimmune forms of pancreatitis, but other glands (e.g. salivary glands and thyroid) can be involved, as well as lungs, kidneys, heart, retroperitoneum, mediastinum and aorta [22].
Infectious Aortitis
Infectious aortitis is an infectious and inflammatory process of the aortic wall directly induced by micro-organisms. In the preantibiotic era, it was most likely a complication of bacterial endocarditis secondary to Streptococcus pyogenes, Streptococcus pneumoniae, and Staphylococcus. Nowadays, the most common pathogens, which account for almost 40 % of infections, include Staphylococcus aureus and Salmonella spp. Other pathogens involved include Treponema pallidum, Mycobacterium tuberculosis (less common today in developed countries), and other bacteria such as Listeria, Bacteroides fragilis, Clostridium septicum, and Campylobacter jejuni [23]. The aorta is normally very resistant to infection; however, an abnormal aortic wall, like that associated with atherosclerotic disease, preexisting aneurysm, medial degeneration, diabetes, vascular/valvular malformation, medical devices, or surgery, makes it more susceptible to infection, if a bacteriaemia occurs [24]. Mechanisms of infection include hematogenous spread (e.g. in non-typhoid Salmonella spp. Gastroenteritis), contiguous seeding from adjacent infection, septic emboli of the aortic vasa vasorum and traumatic or iatrogenic inoculation. Infected (or “mycotic”) aortic aneurysms are part of the spectrum of infectious aortitis and account for <3 % cases of aortic aneurysms. Men are affected more often than women, with most cases seen in adults after the fifth decade of life: elderly or immunocompromised patients are more susceptible to bacterial seeding at the level of preexisting aortic lesions. Both host leukocytes and responsible bacteria can induce aortic wall lesions, namely extracellular matrix degradation, by producing a variety of proteases including matrix metalloprotease-1, −2, −8 and −9 [25]. The collagenase activity may be relatively localized, leading to formation of a saccular abdominal aortic aneurysm or pseudoaneurysm in an otherwise normal appearing vessel. Collagenase activity may also be intensive, which may explain the rapid course associated with infected abdominal aortic aneurysms. Typical pathology findings include aortic atherosclerosis, acute suppurative inflammation, neutrophil infiltration, and bacterial clumps. About two-thirds of patients can show acute inflammation superimposed on severe chronic atherosclerosis; the remainder show atherosclerosis with chronic inflammation or pseudoaneurysms [25].
A Challenging Diagnosis
Clinical Pictures
Since symptoms and signs associated with aortitis during the initial phase of the disease are unspecific, a high level of diagnostic suspicion is required for an early diagnosis and a timely treatment.
Giant cell arteritis usually develops later in life compared to TKA, with only few cases reported at an age younger than 50, and is twice more frequent in women than in men. The clinical onset is quite abrupt so that patients are often able to tell a certain date for the appearance of symptoms. The frequent involvement of the temporal artery leads to the most evident symptoms, i.e. localized headache, scalp tenderness, jaw claudication. Either in association with those symptoms or isolately, the involvement of other vessels, also including the aorta, may cause impairment of vision (anterior ischemic optic neuropathy, amaurosis fugax or diplopia) together with signs of inflammation, fever of unknown origin with night sweats, claudication of the upper limbs, rarely hearing impairment and dizziness, stroke, symptoms of aortic insufficiency and myocardial ischemia [26].
Clinical examination evidences alterations of the temporal artery region, which appears tender, swollen, firm, beaded or reddened with a reduction of the artery pulse. Similar signs can be found in the occipital region as well. At the level of the upper limbs there may be an asymmetry of radial pulses and blood pressure or bruits of the subclavian and axillary region. Symptoms and signs of an associated polymyalgia reumatica can also be present, including reduced range of motion of shoulders, particularly with impaired arm abduction, a reduced internal and external rotation of the hip, tenderness of the upper arms and thighs. When the suspicion of GCA arise, fundoscopy by an experienced investigator may be appropriate, even in patients showing no eye impairment [26].
The diagnosis of GCA relies on clinical, laboratory, and histological criteria as described in the 1990 American College of Rheumatology classification scheme [27] (Table 2.3).
The existence of an atypical pattern of GCA has been described, in which the temporal artery is spared and the disease more consistently affects large arteries, such as aorta. In this case the clinical scenario may be completely different, mostly dominated by systemic symptoms as fever, decline in general wellbeing, laboratory evidence of inflammation and rarely pain in the lower back or abdomen. This makes reaching the clinical diagnosis of GCA with large vessel involvement even later that for temporal arteritis, sometimes only after histology of the intraoperative specimen [28].
Early published data on aortic involvement in patients with GCA were based on the rate of aortic aneurysms diagnosed fortuitously or after acute events (aortic dissection and rupture of an aortic aneurysm). Thus, in retrospective studies, the prevalence of aortitis ranged from 3 to 18 % [29]. Owing to the introduction of new imaging techniques, capable to show aortic involvement before the development of structural abnormalities, rates of aortic involvement ranging between 33 and 45 % have been disclosed.
Aortic involvement is most often localized at the ascending aorta, and occurs quite late during the natural history of GCA (median time from diagnosis 11 years for the thoracic location) most often manifesting as annuloaortic ectasia, determining aortic valve insufficiency or ascending aortic aneurysm. Acute aortic dissection is a possible complication and occasionally represents the first evidence of disease (within 1 year median time of diagnosis). Abdominal aortic aneurysm can also develop and aneurysms are usually present in the thoracic descending segment in the late phase of the disease [30].
Diagnosis of Takayasu arteritis is usually delayed, as a result of the vague nature of the symptoms in its initial phase (often referred to as “pre-pulseless phase”). Mirroring the general systemic inflammation, symptoms of this stage may include fever, malaise, weight loss, night sweat, arthralgia and myalgia [31]. In the late phase the chronic inflammatory process leads to vascular lesions such as aneurismal dilatation, as a consequence of disruption of the connective scaffold in the arterial wall. During the late (“pulseless”) phase, systemic manifestations usually remit significantly and symptoms are mainly related to organ ischemia: arm claudication, dizziness, headache, stroke, visual impairment, renal arterial hypertension, angina, myocardial infarction, pulmonary hypertension [26].
The involvement of the aorta and its main branches is common in this disease, most frequently in the abdominal segment, followed by the descending thoracic aorta and the aortic arch. A 53 % prevalence of aorta stenosis has been reported, in 70 % cases affecting the abdominal aorta [32]. Rapid expansion of aortic aneurysms (45 % cases), aortic rupture (33 %) and (more rarely) intramural hematoma and acute aortic dissection, constitute possible severe complications reported to occur in TKA aortitis [33, 34].
Clinical examination should focus on vascular and neurologic systems: a check of the arterial pulses and auscultation of the subclavian, carotid, abdominal and femoral region may evidence asymmetry of pulses and bruits; a bilateral check of blood pressure in the arms and in the legs should always be performed, as it may show significant pressure differences; a neurological examination may detect signs of an ischemic neurological damage [26].
The onset of specific symptoms and signs of TKA is usually early, i.e. during the third or fourth decade of life. Classification criteria have been developed in 1990 by the American College of Rheumatology for TKA [35] (Table 2.2). The most recently issued classification of TKA, based on the vessels involved, distinguishes: type I, involvement of the main branches from the aortic arch; type IIa, involvement of the ascending aorta, aortic arch and its branches; type IIb, involvement of the ascending aorta, aortic arch and its branches, and thoracic descending aorta; type III, involvement of the thoracic descending aorta, abdominal aorta and/or renal arteries; type IV, involvement of the abdominal aorta and/or renal arteries; and type V, the combined features of type IIb and IV [8].
Table 2.2
Diagnostic criteria for Takayasu’s arteritis (according to the American College of Rheumatology)
Age of 40 years or less at disease onset |
Claudication of the extremities |
Decreased pulsation at one or both brachial arteries (compared to pulses at lower limbs) |
Systolic blood pressure difference of >10 mmHg between the two arms |
Bruit over the subclavean artery or the aorta |
Angiography evidence of focal or segmental occlusion or narrowing of large arteries (including the aorta), not resulting from arteriosclerosis or fibromuscular dysplasia |
Non infectious aortitis may be secondary to rheumatic disease, usually driven by aberrant immune responses, giving rise to clinical pictures in which the specific manifestations of the aortic involvement may be confounded by the systemic clinical scenario dominated by the underlying disease or may initially be overlooked by both patients and physicians.
Ankylosing spondylitis (AS) is part of a group of diseases called spondyloarthropaties, associated with HLA-B27 antigen, characterized by sacroilitis, enthesitis, inflammatory bowel disease or psoriasis. It begins with back pain and stiffness during the second or third decade of life, affecting men two to three times more than women. Diagnosis requires at least four of the following criteria: age younger than 40 at onset, insidious onset of arthropathy, back pain for more than 3 months, morning stiffness, improvement with exercise. Aortitis is present in 80 % of patients with long-standing AS, usually affecting the aortic root and the aortic valve, with insufficiency. AS may also affect the myocardium with impairment of the conduction system [36].
Cogan’s syndrome is a rare disease, characterized by ocular, inner ear, and vascular inflammation. Cardiovascular manifestations include aortitis and necrotizing vasculitis, which may induce coronary, renal, and iliac artery stenosis. About 10 % of patients may have aortitis with aortic aneurysm, and valvulitis with aortic insufficiency. Young male patients are predominantly affected, usually presenting with eye redness, photophobia, or eye pain from interstitial keratitis, audiovestibular manifestations similar to those in Ménière syndrome, neural deafness, and possibly symptoms of aortic insufficiency with or without associated ischemic syndromes due to coronary or iliac stenosis, or hypertension related to renal artery stenosis [37].
Relapsing polychondritis is a paroxysmal and progressive inflammatory disease of the cartilaginous structures, affecting the ear, nose, and hyaline cartilage of the tracheobronchial tree. It is caused by autoimmune response against proteoglycan rich tissues. Aortic involvement may be observed in 5 % of patients, resulting in aneurysm formation in the thoracic and abdominal aorta and obliterans vasculitis in other medium-sized and large arteries. Typical of the acute phases is the histological picture of vasa vasorum extending also through both the media and the edematous intima [38].
Aortitis may be associated also with Behçet’s disease, a systemic chronic disease with typically relapsing course affecting predominantly males of the Mediterranean area and Eastern countries. Its diagnosis is made upon the criteria established by the International Study Group for Behçet’s Disease: presence of oral ulceration and at least two between genital ulceration, eye lesions, skin lesions or a positive pathergy test. In one-fifth of patients affected by aortitic complication, multiple pseudoaneurysms can develop, also involving the iliac, femoral, popliteal, and subclavian arteries.
Less frequently, aortitis may be associated with other rheumatologic diseases including rheumatoid arthritis (5 %), Reiter disease (<1 %) and systemic lupus erythematosus (few cases reported).
No specific clinical picture is associated with idiopathic aortitis of the thoracic segment, which can indeed be asymptomatic and detected incidentally: diagnosis is made in such cases at the time of histopathology review after thoracic aortic aneurysm surgery. In some cases unspecific thoracic pain can occur, but in most instances no systemic inflammatory symptoms are present. An idiopathic inflammatory aneurysm of the abdominal aorta can present with back or abdominal pain and constitutional symptoms, similarly to other non-infectious etiologies, and differentiation can be suggested by laboratory results and by histological analysis after surgical excision. Clinical onset of retroperitoneal fibrosis can be accompanied by renal function impairment, due to ureteral obstruction and in some cases by intestinal symptoms (e.g. abdominal pain and/or mass with or without sickness and vomit, related to duodenal obstruction) [39].
Infectious aortitis is a severe clinical entity, insidious insofar as it can be virtually undistinguished from non-infectious forms in terms of clinical presentation, and associated with a high inherent risk of acute and life-threatening complications. Salmonella spp. are reported to be the commonest pathogens involved in infective aortitis, accounting for almost 40 % of infective aortitis together with Staphylococcus aureus, mostly involving the abdominal aorta. The more frequent route of infection is a bacteremia following an ingestion of contaminated food, and a subsequent colonization of a pre-existing aortic atherosclerotic lesion. Aortic infection from a contiguous site, such as a paravertebral abscess complicating a spondylodiscitis is less common. Rare complications are aorto-enteric fistula and endo-myocardial abscess. The natural history of infectious aortitis is characterized by the progressive expansion of the aneurysm, with a greater tendency to rupture, compared to other etiologies, if not diagnosed and treated promptly. The majority of patients affected by infective aortitis are symptomatic, especially in the aneurysmal stage of the disease. Fever and back pain are the most common symptoms, being present respectively in the 77 % and 65 % of patients with infected aortic aneurysm. Chills, sweats, abdominal symptoms as nausea and vomiting are other possible symptoms [40].
Pneumoccoccal aortitis is rare and is usually due to bacteriemic spread from distant infection foci, such as pneumonia, urinary tract infections, endocarditis, osteomyelitis, cellulitis. Abdominal aorta is the segment most often involved by pneumococcal aortitis, followed by descending thoracic aorta [41].
Aortitis may be a clinical consequence of Treponema pallidum determining obliterative vasculitis of aortic vasa vasorum, during the third (late) phase of syphilis. After a progressive decrease in its epidemiological importance over the last century, primary syphilis has doubled its incidence during the first decade of the new century, with a majority of cases among homosexual men. This probably heralds a new resurgence of tertiary syphilis, in the next years, with a new epidemiological pattern of infective aortitis. Luetic aortitis typically involves the tubular portion of the ascending aorta, aortic arch and descending thoracic aorta, sparing the sinuses of Valsalva. Consequently, aortic insufficiency associated to aortic root dilatation has been only seldom reported. Clinical diagnosis is most often made based upon serologic confirmation of syphilis and a characteristic pattern of vascular involvement [42].
Other microorganisms, such as Enterococcus spp., Listeria monocytogenes, Bacteroides fragilis, Clostridium septicum, human immunodeficiency virus (HIV), Mycobacterium tuberculosis may less frequently cause infectious aortitis. A positive history for signs and symptoms of the primary infection should guide the diagnosis towards infectious etiology, if an aortitis has been detected, importantly distinguishing it from autoimmune etiology. The warning has been issued that tuberculous aortitis, possibly evolving towards vessel stenosis or occlusion, might be misdiagnosed as Takayasu arteritis and erroneously treated by glucocorticoids, which may obviously worsen the infection course.
Imaging and Laboratory
The relevant, though complementary role played by imaging in the diagnosis of aortitis was officially recognized in the 2010 American College of Cardiology / American Heart Association Guidelines for the diagnosis and management of patients with thoracic aortic disease (Class I, level C), as it was in the American College of Rheumatology criteria for the diagnosis of Takayasu arteritis [35, 43].
Imaging provides important information for establishing the diagnosis, contributing in the differentiation between aortitis and other causes of aortic dilatation or large vessel stenosis, estimating the extent of disease, helping to monitor disease activity and response to therapy, and guiding biopsies (in GCA-associated temporal arteritis). The different available imaging methods are used to describe, with different specificity, the two elements of (1) the aortic lumen and (2) aortic wall changes. In large-vessel vasculitis, imaging studies document the anatomic distribution of the lesion, characterized by homogeneous artery wall swelling and aortic dilatation or peculiar large vessel stenoses with smoothly tapered luminal narrowing.
Giant cell arteritis typically involves the branches of the external carotid arteries. The aorta and its main branches are usually unaffected but the possible occurrence of an atypical pattern of large-vessel GCA with negative temporal artery biopsy is reported in up to 25 % of patients [26] and often unsuspected until life-threatening complications occur. Large-vessel form of GCA usually involves the axillary arteries bilaterally and less frequently the subclavian, brachial, femoro-popliteal axis or the aorta itself. However, because aortitis-related complications may be a source of both severe morbidity and mortality, routine screening for aortic involvement is mandatory in patients with any form of GCA [28].
The peripheral aortic branches are easily accessible to ultrasonography (US) that shows a perivascular hypoechoic halo similar to the finding at the temporal artery (the “halo sign”), which reflects wall edema. Soon after pharmacologic treatment initiation, wall edema decreases and US reveals an increase of the wall echogenicity because fibrosis occurs, being still visible in more than a half of patients even after 1 year of treatment [44]. When GCA affects arteries in the lower limbs, special attention must be paid for a differential diagnosis with atherosclerosis that often occurs at these sites but with different characteristics (atherosclerotic plaques are usually calcified, asymmetric and inhomogeneous) [45].
Computed tomography (CT) angiography is commonly the initial imaging study performed because it is diffusely available. It has an excellent spatial resolution and multidetector scanners allow multiplanar reformation and three-dimensional reconstruction. Actually, CT is less sensitive than other techniques, as magnetic resonance imaging (MRI) or positron-emission tomography (PET), for identifying early wall changes but in an advanced phase it is useful to reveal luminal changes such as stenosis, occlusion, dilatation, aneurysm, calcification and mural thrombi. Contrast-enhanced CT scan may help diagnosis of aortitis showing a concentric thickening (>3 mm) of the arterial wall with post-contrast enhancement [28].
MRI can provide accurate information on involvement of the aorta and its branches, moreover high resolution MRI can investigate temporal arteries. MRI is able to detect the earliest vascular inflammation in the vessel wall and also the luminal changes of the mature phase: findings in GCA include circumferential thickening of the vessel-wall in T1-weighted images, producing a high signal on T2-weighted images (wall edema), and post-gadolinium enhancement in the affected segment [28].
18-Fluorodeoxyglucose (18FDG) – PET-CT is a useful imaging modality in the assessment of active inflammation in cardiovascular diseases including aortitis, atherosclerosis and acute dissection. Normally there is no radiotracer accumulation in the arterial wall, thus any 18FDG up-take can be consider a sign of inflammatory infiltrates or infection (Fig. 2.2). Large-vessel FDG uptake is usually graded on a 4-point scale: none (grade 0), lower than liver uptake (grade 1), similar to liver uptake (grade 2) and higher than liver uptake (grade 3). Grades 2–3 are relatively specific for vasculitis, while grade 1, or rarely 2, has been observed in atherosclerotic vessels [46]. Moreover, to exclude false positivity due to atherosclerosis (especially in lower limb lesions), some authors suggest relying only on the upper-body sites of 18-FDG uptake for the diagnosis of GCA [47]. In GCA 18FDG-PET may reveal early inflammation sites even in the absence of detectable structural changes at CT: abnormal uptake in the aortic arch or large thoracic arteries is found in more than 50 % of affected patients. This is of particular importance for establishing the diagnosis in atypical clinical scenarios with predominance of systemic signs of inflammation, with negative temporal artery biopsy [28]. Since inflammatory cell infiltration is likely to happen prior to the development of wall edema, PET can be even more sensitive for early aortitis than MRI. Contrary to MRI, PET cannot investigate cranial arteries because of its low spatial resolution and the background noise derived from the brain (high FDG uptake of the neuronal cells). PET may also be useful for monitoring the response to treatment: the persistence of 18FDG uptake in the arterial wall at follow-up despite an adequate therapy has been described to have a predictive role for vascular remodeling and aneurismal dilatation [48].
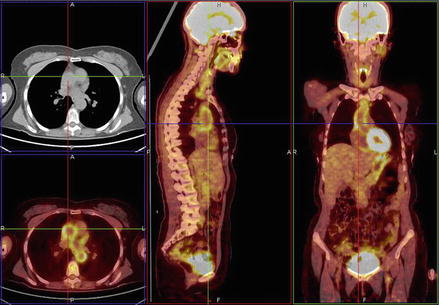
Figure 2.2
18FDG-PET-CT showing hyper-uptake at the level of the ascending aorta. Such levels of uptake are highly indicative of aortitis; lower levels may be associated with atherosclerotic lesions, whereas the normal has no detectable uptake (Courtesy of Drs M. Bifulco and F. Porcaro, Nuclear Medicine Diagnostics Unit of the Monaldi Hospital, Naples, Italy)
No specific laboratory tests exist for the diagnosis of GCA. Erythrocyte Sedimentation Rate (ESR) and also C Reactive Protein (CRP) readings are high in most patients, and ESR elevation is included between the diagnostic criteria, although up to 10 % of patients with documented GCA have normal sedimentation rates at the time of diagnosis. On the contrary, an elevated ESR can be seen in most of the disorders usually considered in the differential diagnosis of patients with possible vasculitis, notably infections and malignancies, thus limiting the diagnostic usefulness of this test. Acute phase reactants may serve as simple tools for monitoring disease activity during therapy. Many patients have mild to moderate anemia, thrombocytosis and slightly elevated transaminases [26, 43].
While large vessel GCA typically involves the axillary arteries, in Takayasu’s arteritis the most commonly involved sites are the subclavian arteries (93 %), followed by the aorta (65 %), and the common carotid arteries (58 %) [26]. Other possible sites described for TKA are renal, vertebral, innominate, axillary, superior mesenteric, common iliac, and pulmonary arteries.
Differently from GCA, CT angiography is essential in the early steps of the diagnostic process for TKA, being the imaging technique with the highest predictive power for demonstrating the abnormalities of the affected vessels. The typical finding in the early stage of the disease is the wall thickening that has been described as the “double ring” sign. This is due to edema in the intimal layer which gives a low-density signal next to a high-density signal from the infiltrated media and adventitia. In the chronic phase of the disease (≥5 years) CT scan may show calcifications of the previous inflamed sites: these are commonly linear and tend to spare the ascending aorta [21].
MRI may also help in the diagnosis of TKA because of its intrinsic ability to investigate the early wall changes occurring before lumen stenosis develops, with findings similar to those in large-vessel GCA. Phase-contrast (PC) – MRI and magnetic resonance angiography (MRA) can also document multiple stenoses, mural thrombi, thickening of aortic valve cusps, and pericardial effusions [21, 49]. For the absence of ionizing radiations, MRI is recommended for serial imaging follow-up especially in young patients.
In a normal carotid wall, US shows an hypoechoic space known as the intima-media complex (IMC), in between two hyperechogenic layers. When edema occurs in the arterial wall, there is an increased and diffuse thickening of the IMC that has been referred to as “macaroni sign”, unique of TKA [50]. This diffuse thickening, together with the arterial segments involved, help to differentiate vasculitis from atherosclerosis. The stenosing lesions evolve quite slowly, which explains the common presence of collaterals, with reported cases of reverse flow in vertebral arteries with or without the subclavian steal phenomenon in patients with Takayasu disease [51]. US is useful in TKA also for the investigation of the aortic valve, ascending aorta and pulmonary artery [52].
18FDG-PET represents a promising, yet not definitively established, method to help in the diagnosis of TKA in patients with constitutional symptoms and fever of unknown origin. Hybrid imaging with 18FDG-PET (detecting circumferential increased metabolic activity) and CT or MRI (allowing more precise anatomic localization of the disease) has emerged as a valuable tool in diagnosing and monitoring treatment response in TKA aortitis. The European League Against Rheumatism (EULAR) recommends PET, together with MRI, for diagnosing large-vessel vasculitis, most notably in patients with TKA, as histological documentation is difficult to obtain in the large-vessel forms of the disease [53].
In TKA laboratory test findings are similar to those of GCA. The ESR and CRP are in most cases highly elevated in active disease, although a smaller number of patients have normal ESR or CRP values [26].
When non-infectious aortitis is ascertained, in the absence of an identifiable vasculitis or rheumatic systemic syndrome with possible secondary aortic involvement, idiopathic aortitis is diagnosed. Usually the diagnosis is made post-operatively on the basis of the histological findings on the aortic specimen (giant cells or lymphoplasmacytic inflammation). Patients with idiopathic aortitis have more diffuse and more often extensive (also thoracic descending and thoraco-abdominal) dilatation of the aorta compared with those with non-inflammatory dilatations [54]. Idiopathic aortitis patients are generally older at presentation and have greater diameters than those with large vessel vasculitis-associated aortitis, probably related to the silently progressing nature of the disease [55]. CT-scan identifies the inflammatory aneurysm as a hypo-dense mass with thickening of the periaortic tissues that show delayed contrast enhancement in CT angiography following the rapid intra-luminal enhancement.
In idiopathic inflammatory abdominal aneurysms the thickening of the aortic wall/periaortic tissues typically spares the posterior aspect of the vessel [56]. CT is also important to asses possible adhesions of the mass with the abdominal organs in order to plan the surgical strategy (i.e. transperitoneal versus retroperitoneal approach). In the pre-operative phase, MRI helps detailing aneurysm localization (suprarenal versus infrarenal) and demonstrates the presence of periaortic inflammation, adventitial thickening and turbulent flow inside the aneurysm. Diffusion-weighted MR imaging shows a hyperintense halo surrounding the aneurysm.
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


