Figure 5-1. Qualitative and semi-quantitative representation of receptor binding properties. Throughout this chapter, the receptor binding properties of the atypical antipsychotics are represented both graphically and semi-quantitatively. Each drug is represented as a blue sphere, with its most potent binding properties depicted along the outer edge of the sphere. Additionally, each drug has a series of colored boxes associated with it. Each colored box represents a different binding property, and binding strength is indicated by the size of the box and the number of plus signs. Within the colored box series for any particular antipsychotic, larger boxes with more plus signs (positioned to the left) indicate stronger binding affinity, while smaller boxes with fewer plus signs (positioned to the right) represent weaker binding affinity. The series of boxes associated with each drug are arranged such that the size and positioning of a box reflect the binding potency for a particular receptor. The vertical dotted line cuts through the dopamine 2 (D2) receptor binding box, with binding properties that are more potent than D2 on the left and those that are less potent than D2 on the right. All binding properties are based on the mean values of published Ki (binding affinity) data (http://pdsp.med.unc.edu). The semi-quantitative depiction used throughout this chapter provides a quick visual reference of how strongly a particular drug binds to a particular receptor. It also allows for easy comparison of a drug’s binding properties with those of other atypical antipsychotics.
Conventional antipsychotics
What makes an antipsychotic “conventional”?
In this section we will discuss the pharmacologic properties of the first drugs that were proven to effectively treat schizophrenia. A list of many conventional antipsychotic drugs is given in Table 5-1. These drugs are usually called conventional antipsychotics, but they are sometimes also called classical antipsychotics, or typical antipsychotics, or first-generation antipsychotics. The earliest effective treatments for schizophrenia and other psychotic illnesses arose from serendipitous clinical observations more than 60 years ago, rather than from scientific knowledge of the neurobiological basis of psychosis, or of the mechanism of action of effective antipsychotic agents. Thus, the first antipsychotic drugs were discovered by accident in the 1950s when a drug with antihistamine properties (chlorpromazine) was serendipitously observed to have antipsychotic effects when this putative antihistamine was tested in schizophrenia patients. Chlorpromazine indeed has antihistaminic activity, but its therapeutic actions in schizophrenia are not mediated by this property. Once chlorpromazine was observed to be an effective antipsychotic agent, it was tested experimentally to uncover its mechanism of antipsychotic action.
| Generic name | Trade name | Comment |
|---|---|---|
| Chlorpromazine | Thorazine | Low potency |
| Cyamemazine | Tercian | Atypical at low doses; popular in France; not available in the US |
| Flupenthixol | Depixol | Depot; not available in the US |
| Fluphenazine | Prolixin | High potency; depot |
| Haloperidol | Haldol | High potency; depot |
| Loxapine | Loxitane | Atypical at low doses |
| Mesoridazine | Serentil | Low potency; QTc issues; second line |
| Perphenazine | Trilafon | High potency |
| Pimozide | Orap | High potency; Tourette’s syndrome; QTc issues; second line |
| Pipothiazine | Piportil | Depot; not available in the US |
| Sulpiride | Dolmatil | May have some atypical properties; not available in the US |
| Thioridazine | Mellaril | Low potency; QTc issues; second line |
| Thiothixene | Navane | High potency |
| Trifluoperazine | Stelazine | High potency |
| Zuclopenthixol | Clopixol | Depot; not available in the US |
Early in the testing process, chlorpromazine and other antipsychotic agents were all found to cause “neurolepsis,” known as an extreme form of slowness or absence of motor movements as well as behavioral indifference in experimental animals. The original antipsychotics were first discovered largely by their ability to produce this effect in experimental animals, and are thus sometimes called “neuroleptics.” A human counterpart of neurolepsis is also caused by these original (i.e., conventional) antipsychotic drugs and is characterized by psychomotor slowing, emotional quieting, and affective indifference.
D2 receptor antagonism makes an antipsychotic conventional
By the 1970s it was widely recognized that the key pharmacologic property of all “neuroleptics” with antipsychotic properties was their ability to block dopamine D2 receptors (Figure 5-2). This action has proven to be responsible not only for the antipsychotic efficacy of conventional antipsychotic drugs, but also for most of their undesirable side effects, including “neurolepsis.”
Figure 5-2. D2 antagonist. Conventional antipsychotics, also called first-generation antipsychotics or typical antipsychotics, share the primary pharmacological property of D2 antagonism, which is responsible not only for their antipsychotic efficacy but also for many of their side effects. Shown here is an icon representing this single pharmacological action.
The therapeutic actions of conventional antipsychotic drugs are hypothetically due to blockade of D2 receptors specifically in the mesolimbic dopamine pathway (Figure 5-3). This has the effect of reducing the hyperactivity in this pathway that is postulated to cause the positive symptoms of psychosis, as discussed in Chapter 4 (Figures 4-12 and 4-13). All conventional antipsychotics reduce positive psychotic symptoms about equally well in schizophrenia patients studied in large multicenter trials if they are dosed to block a substantial number of D2 receptors there (Figure 5-4). Unfortunately, in order to block adequate numbers of D2 receptors in the mesolimbic dopamine pathway to quell positive symptoms, one must simultaneously block the same number of D2 receptors throughout the brain, and this causes undesirable side effects as a “high cost of doing business” with conventional antipsychotics (Figures 5-5 through 5-8). Although modern neuroimaging techniques are able to measure directly the blockade of D2 receptors in the dorsal (motor) striatum of the nigrostriatal pathway, as shown in Figure 5-4, for conventional antipsychotics it is assumed that the same number of D2 receptors is blocked in all brain areas, including the ventral limbic area of striatum known as the nucleus accumbens of the mesolimbic dopamine pathway, the prefrontal cortex of the mesocortical dopamine pathway, and the pituitary gland of the tuberoinfundibular dopamine pathway.
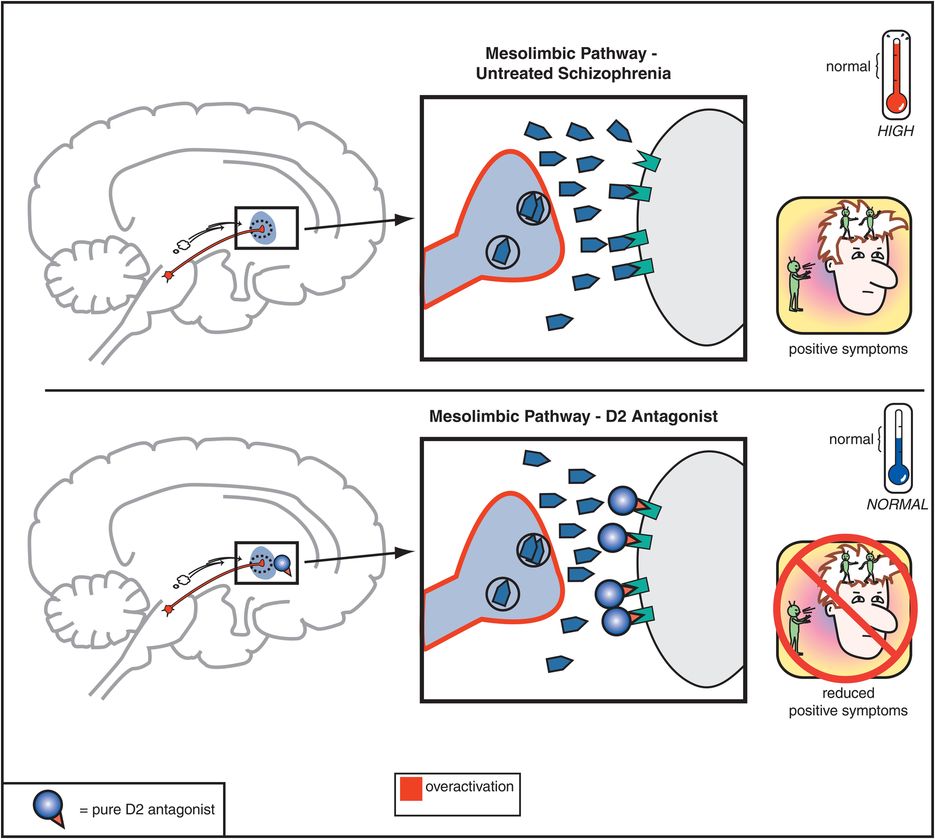
Figure 5-3. Mesolimbic dopamine pathway and D2 antagonists. In untreated schizophrenia, the mesolimbic dopamine pathway is hypothesized to be hyperactive, indicated here by the pathway appearing red as well as by the excess dopamine in the synapse. This leads to positive symptoms such as delusions and hallucinations. Administration of a D2 antagonist, such as a conventional antipsychotic, blocks dopamine from binding to the D2 receptor, which reduces hyperactivity in this pathway and thereby reduces positive symptoms as well.
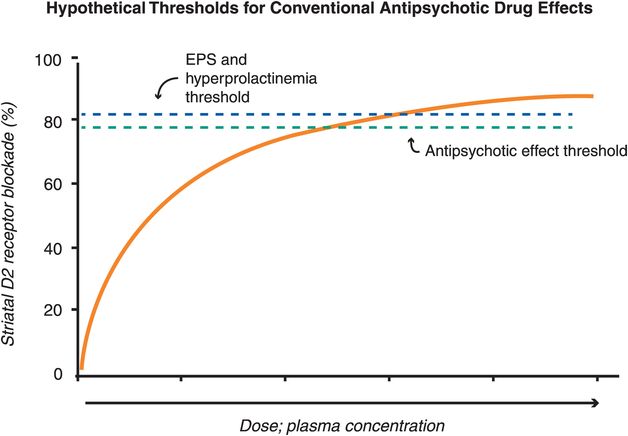
Figure 5-4. Hypothetical thresholds for conventional antipsychotic drug effects. All known antipsychotics bind to the dopamine 2 receptor, with the degree of binding determining whether one experiences therapeutic and/or side effects. For most conventional antipsychotics, the degree of D2 receptor binding in the mesolimbic pathway needed for antipsychotic effects is close to 80%, while D2 receptor occupancy greater than 80% in the dorsal striatum is associated with extrapyramidal side effects (EPS) and in the pituitary is associated with hyperprolactinemia. For conventional antipsychotics (i.e,. pure D2 antagonists) it is assumed that the same number of D2 receptors is blocked in all brain areas. Thus, there is a narrow window between the threshold for antipsychotic efficacy and that for side effects in terms of D2 binding.
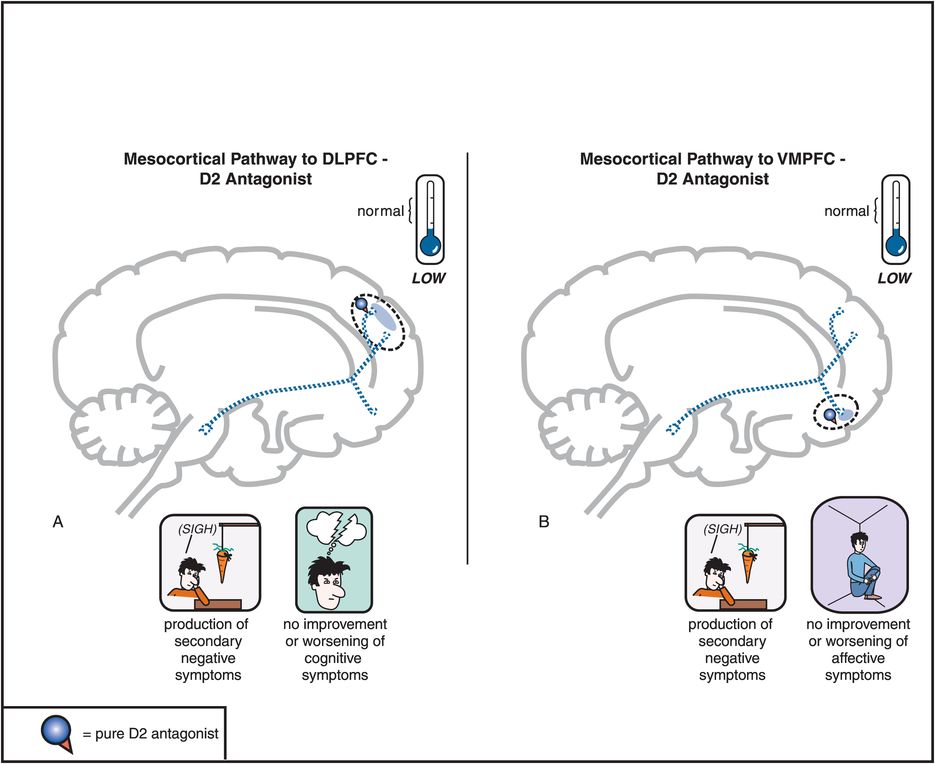
Figure 5-5. Mesocortical dopamine pathway and D2 antagonists. In untreated schizophrenia, the mesocortical dopamine pathways to dorsolateral prefrontal cortex (DLPFC) and to ventromedial prefrontal cortex (VMPFC) are hypothesized to be hypoactive, indicated here by the dotted outlines of the pathway. This hypoactivity is related to cognitive symptoms (in the DLPFC), negative symptoms (in the DLPFC and VMPFC), and affective symptoms of schizophrenia (in the VMPFC). Administration of a D2 antagonist could further reduce activity in this pathway and thus not only not improve such symptoms but actually potentially worsen them.
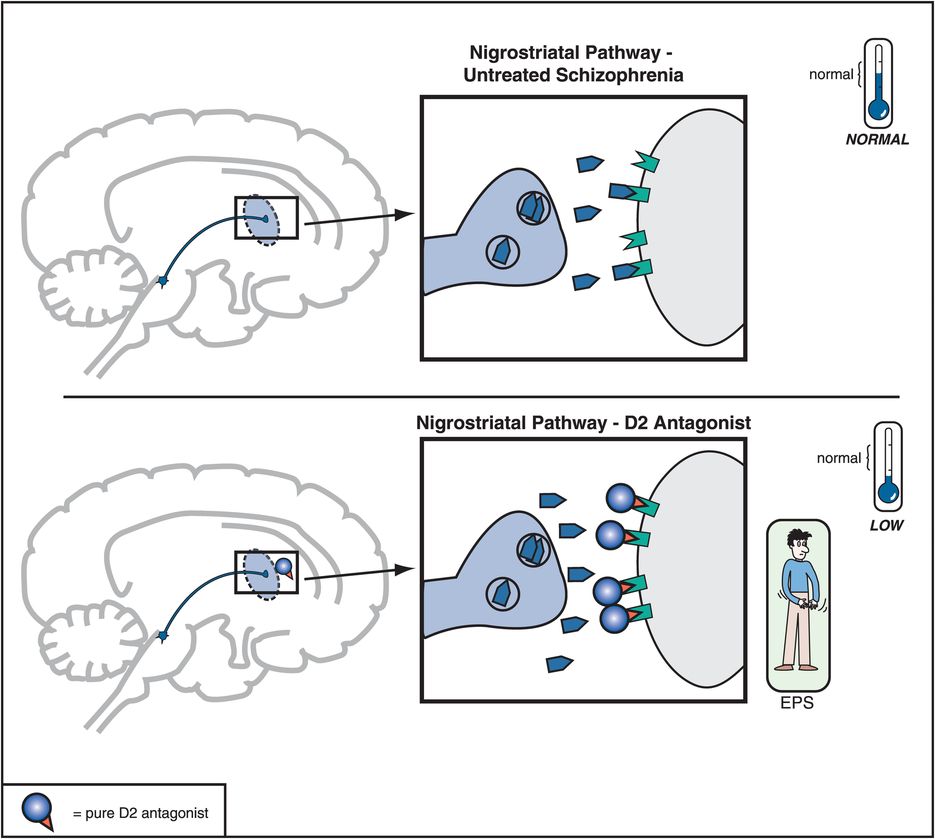
Figure 5-6. Nigrostriatal dopamine pathway and D2 antagonists. The nigrostriatal dopamine pathway is theoretically unaffected in untreated schizophrenia. However, blockade of D2 receptors, as with a conventional antipsychotic, prevents dopamine from binding there and can cause motor side effects that are often collectively termed extrapyramidal symptoms (EPS).
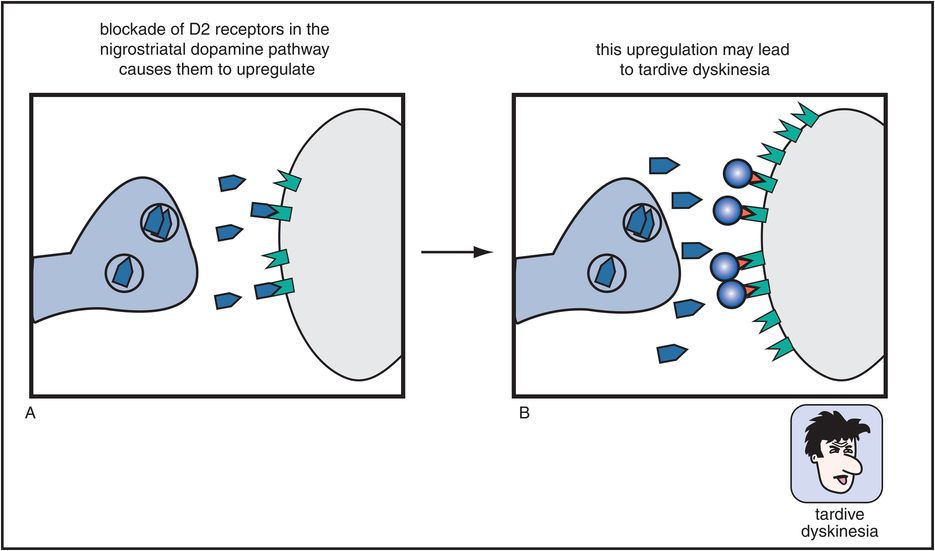
Figure 5-7. Tardive dyskinesia. Long-term blockade of D2 receptors in the nigrostriatal dopamine pathway can cause upregulation of those receptors, which may lead to a hyperkinetic motor condition known as tardive dyskinesia, characterized by facial and tongue movements (e.g., tongue protrusions, facial grimaces, chewing) as well as quick, jerky limb movements. This upregulation may be the consequence of the neuron’s futile attempt to overcome drug-induced blockade of its dopamine receptors.
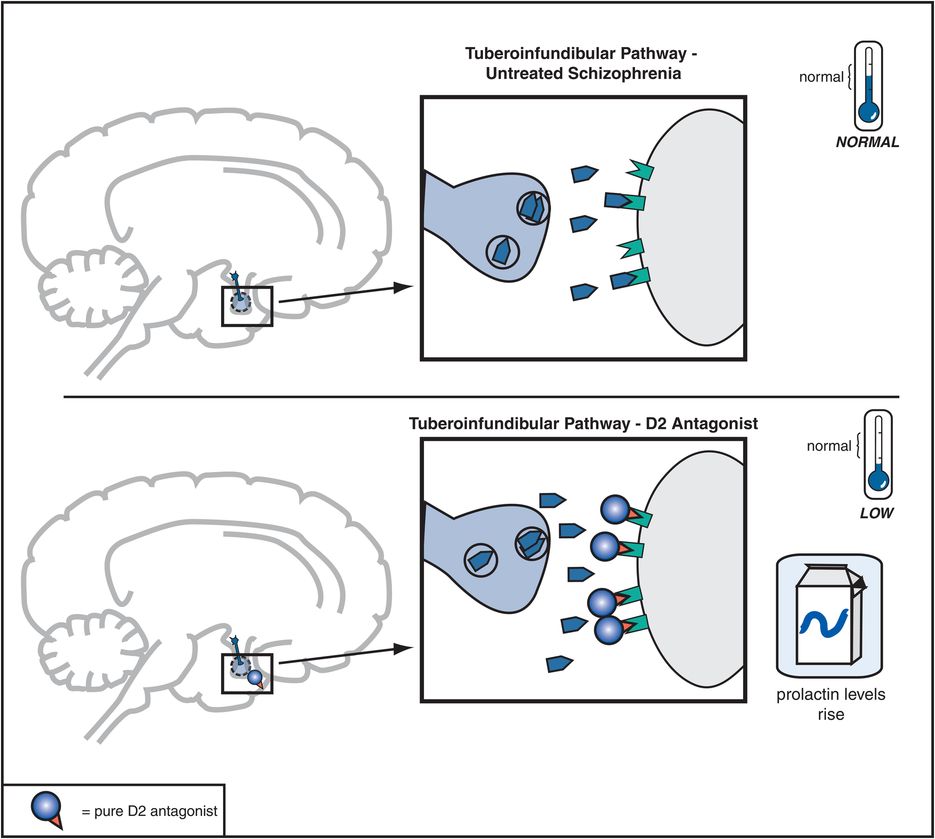
Figure 5-8. Tuberoinfundibular dopamine pathway and D2 antagonists. The tuberoinfundibular dopamine pathway, which projects from the hypothalamus to the pituitary gland, is theoretically “normal” in untreated schizophrenia. D2 antagonists reduce activity in this pathway by preventing dopamine from binding to D2 receptors. This causes prolactin levels to rise, which is associated with side effects such as galactorrhea (breast secretions) and amenorrhea (irregular menstrual periods).
Neurolepsis
D2 receptors in the mesolimbic dopamine system are postulated to mediate not only the positive symptoms of psychosis, but also the normal reward system of the brain, and the nucleus accumbens is widely considered to be the “pleasure center” of the brain. It may be the final common pathway of all reward and reinforcement, including not only normal reward (such as the pleasure of eating good food, orgasm, listening to music) but also the artificial reward of substance abuse. If D2 receptors are stimulated in some parts of the mesolimbic pathway, this can lead to the experience of pleasure. Thus, if D2 receptors in the mesolimbic system are blocked, this may not only reduce positive symptoms of schizophrenia, but also block reward mechanisms, leaving patients apathetic, anhedonic, lacking motivation, interest, and joy from social interactions, a state very similar to that of negative symptoms of schizophrenia. The near shutdown of the mesolimbic dopamine pathway necessary to improve the positive symptoms of psychosis (Figure 5-4) may contribute to worsening of anhedonia, apathy, and negative symptoms, and this may be a partial explanation for the high incidence of smoking and drug abuse in schizophrenia.
Antipsychotics also block D2 receptors in the mesocortical DA pathway (Figure 5-5), where DA may already be deficient in schizophrenia (see Figures 4-14 through 4-16). This can cause or worsen negative and cognitive symptoms even though there is only a low density of D2 receptors in the cortex. An adverse behavioral state can be produced by conventional antipsychotics, and is sometimes called the “neuroleptic-induced deficit syndrome” because it looks so much like the negative symptoms produced by schizophrenia itself, and is reminiscent of “neurolepsis” in animals.
Extrapyramidal symptoms and tardive dyskinesia
When a substantial number of D2 receptors are blocked in the nigrostriatal DA pathway, this will produce various disorders of movement that can appear very much like those in Parkinson’s disease; this is why these movements are sometimes called drug-induced parkinsonism. Since the nigrostriatal pathway is part of the extrapyramidal nervous system, these motor side effects associated with blocking D2 receptors in this part of the brain are sometimes also called extrapyramidal symptoms, or EPS (Figures 5-4 and 5-6).
Worse yet, if these D2 receptors in the nigrostriatal DA pathway are blocked chronically (Figure 5-7), they can produce a hyperkinetic movement disorder known as tardive dyskinesia. This movement disorder causes facial and tongue movements, such as constant chewing, tongue protrusions, facial grimacing, and also limb movements that can be quick, jerky, or choreiform (dancing). Tardive dyskinesia is thus caused by long-term administration of conventional antipsychotics and is thought to be mediated by changes, sometimes irreversible, in the D2 receptors of the nigrostriatal DA pathway. Specifically, these receptors are hypothesized to become supersensitive or to “upregulate” (i.e., increase in number), perhaps in a futile attempt to overcome drug-induced blockade of D2 receptors in the striatum (Figure 5-7).
About 5% of patients maintained on conventional antipsychotics will develop tardive dyskinesia every year (i.e., about 25% of patients by 5 years), not a very encouraging prospect for a lifelong illness starting in the early twenties. The risk of developing tardive dyskinesia in elderly subjects may be as high as 25% within the first year of exposure to conventional antipsychotics. However, if D2 receptor blockade is removed early enough, tardive dyskinesia may reverse. This reversal is theoretically due to a “resetting” of these D2 receptors by an appropriate decrease in the number or sensitivity of them in the nigrostriatal pathway once the antipsychotic drug that had been blocking these receptors is removed. However, after long-term treatment, the D2 receptors apparently cannot or do not reset back to normal, even when conventional antipsychotic drugs are discontinued. This leads to tardive dyskinesia that is irreversible, continuing whether conventional antipsychotic drugs are administered or not.
Is there any way to predict those who will be harmed with the development of tardive dyskinesia after chronic treatment with conventional antipsychotics? Patients who develop EPS early in treatment may be twice as likely to develop tardive dyskinesia if treatment with a conventional antipsychotic is continued chronically. Also, specific genotypes of dopamine receptors may confer important genetic risk factors for developing tardive dyskinesia with chronic treatment using a conventional antipsychotic. Risk of new cases of tardive dyskinesia, however, can diminish considerably after 15 years of treatment, presumably because patients who have not developed tardive dyskinesia despite 15 years of treatment with a conventional antipsychotic have lower genetic risk factors for it.
A rare but potentially fatal complication called the “neuroleptic malignant syndrome,” associated with extreme muscular rigidity, high fevers, coma, and even death, and possibly related in part to D2 receptor blockade in the nigrostriatal pathway, can also occur with conventional antipsychotic agents.
Prolactin elevation
Dopamine D2 receptors in the tuberoinfundibular DA pathway are also blocked by conventional antipsychotics, and this causes plasma prolactin concentrations to rise, a condition called hyperprolactinemia (Figure 5-8). This is associated with conditions called galactorrhea (i.e., breast secretions) and amenorrhea (i.e., irregular or lack of menstrual periods). Hyperprolactinemia may thus interfere with fertility, especially in women. Hyperprolactinemia might lead to more rapid demineralization of bones, especially in postmenopausal women who are not taking estrogen replacement therapy. Other possible problems associated with elevated prolactin levels may include sexual dysfunction and weight gain, although the role of prolactin in causing such problems is not clear.
The dilemma of blocking D2 dopamine receptors in all dopamine pathways
It should now be obvious that the use of conventional antipsychotic drugs presents a powerful dilemma. That is, there is no doubt that conventional antipsychotic medications exert dramatic therapeutic actions upon positive symptoms of schizophrenia by blocking hyperactive dopamine neurons in the mesolimbic dopamine pathway. However, there are several dopamine pathways in the brain, and it appears that blocking dopamine receptors in only one of them is useful (Figure 5-3), whereas blocking dopamine receptors in the remaining pathways may be harmful (Figures 5-4 through 5-8). The pharmacologic quandary here is what to do if one wishes simultaneously to decrease dopamine in the mesolimbic dopamine pathway in order to treat positive psychotic symptoms theoretically mediated by hyperactive mesolimbic dopamine neurons and yet increase dopamine in the mesocortical dopamine pathway to treat negative and cognitive symptoms, while leaving dopaminergic tone unchanged in both the nigrostriatal and tuberoinfundibular dopamine pathways to avoid side effects. This dilemma may have been addressed in part by the atypical antipsychotic drugs described in the following sections, and is one of the reasons why the atypical antipsychotics have largely replaced conventional antipsychotic agents in the treatment of schizophrenia and other psychotic disorders throughout the world.
Muscarinic cholinergic blocking properties of conventional antipsychotics
In addition to blocking D2 receptors in all dopamine pathways (Figures 5-3 through 5-8), conventional antipsychotics have other important pharmacologic properties (Figure 5-9). One particularly important pharmacologic action of some conventional antipsychotics is their ability to block muscarinic M1-cholinergic receptors (Figures 5-9 through 5-11). This can cause undesirable side effects such as dry mouth, blurred vision, constipation, and cognitive blunting (Figure 5-10). Differing degrees of muscarinic cholinergic blockade may also explain why some conventional antipsychotics have a lesser propensity to produce extrapyramidal side effects (EPS) than others. That is, those conventional antipsychotics that cause more EPS are the agents that have only weak anticholinergic properties, whereas those conventional antipsychotics that cause fewer EPS are the agents that have stronger anticholinergic properties.
Figure 5-9. Conventional antipsychotic. Shown here is an icon representing a conventional antipsychotic drug. Conventional antipsychotics have pharmacological properties in addition to dopamine D2 antagonism. The receptor profiles differ for each agent, contributing to divergent side-effect profiles. However, some important characteristics that multiple agents share are the ability to block muscarinic cholinergic receptors, histamine H1 receptors, and/or α1-adrenergic receptors.
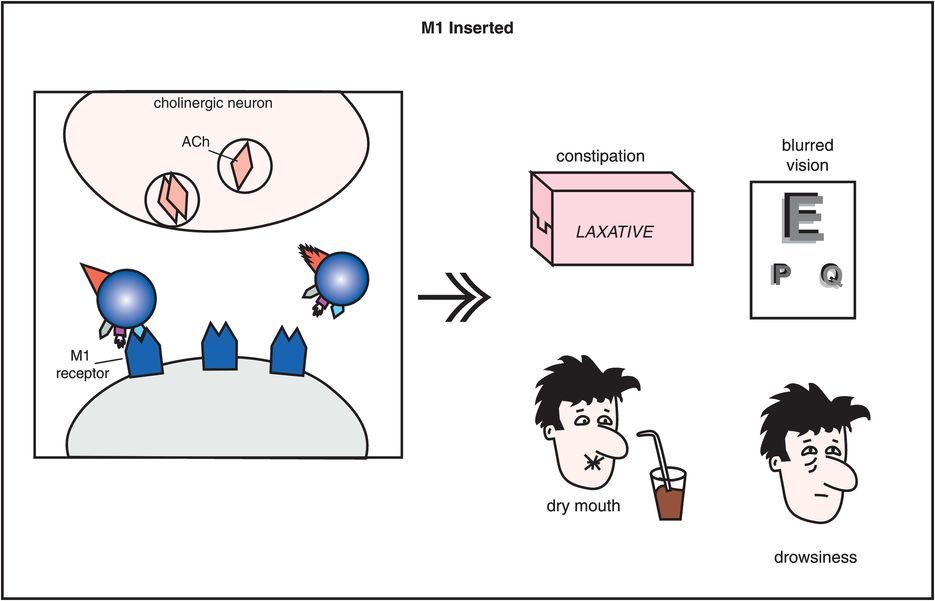
Figure 5-10. Side effects of muscarinic cholinergic receptor blockade. In this diagram, the icon of a conventional antipsychotic drug is shown with its M1 anticholinergic/antimuscarinic portion inserted into acetylcholine receptors, causing the side effects of constipation, blurred vision, dry mouth, and drowsiness.
How does muscarinic cholinergic receptor blockade reduce the EPS caused by dopamine D2 receptor blockade in the nigrostriatal pathway? The reason seems to be based on the fact that dopamine and acetylcholine have a reciprocal relationship with each other in the nigrostriatal pathway (Figure 5-11). Dopamine neurons in the nigrostriatal dopamine pathway make postsynaptic connections with cholinergic neurons (Figure 5-11A). Dopamine normally inhibits acetylcholine release from postsynaptic nigrostriatal cholinergic neurons, thus suppressing acetylcholine activity there (Figure 5-11A). If dopamine can no longer suppress acetylcholine release because dopamine receptors are being blocked by a conventional antipsychotic drug, then acetylcholine becomes overly active (Figure 5-11B).
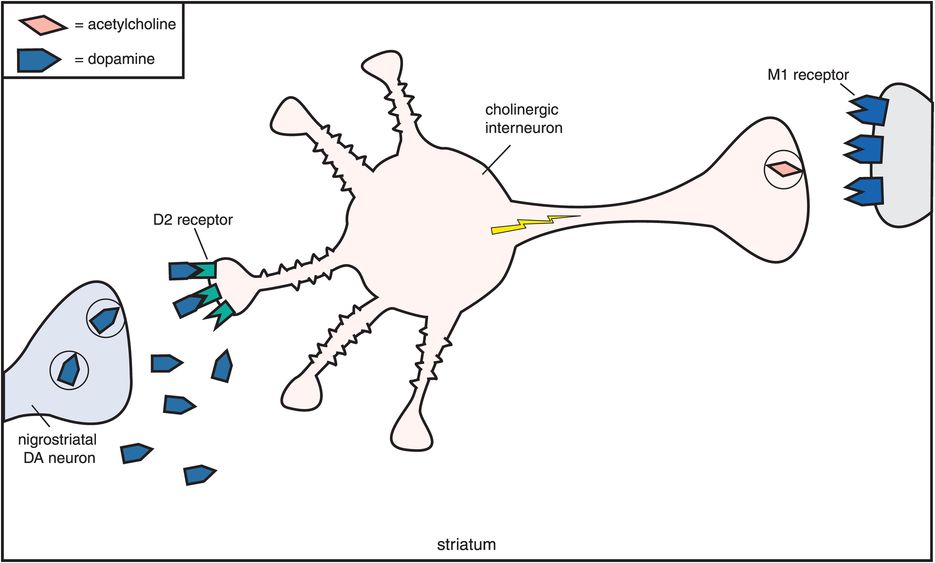
A. Reciprocal relationship of dopamine and acetylcholine. Dopamine and acetylcholine have a reciprocal relationship in the nigrostriatal dopamine pathway. Dopamine neurons here make postsynaptic connections with the dendrite of a cholinergic neuron. Normally, dopamine suppresses acetylcholine activity (no acetylcholine being released from the cholinergic axon on the right).
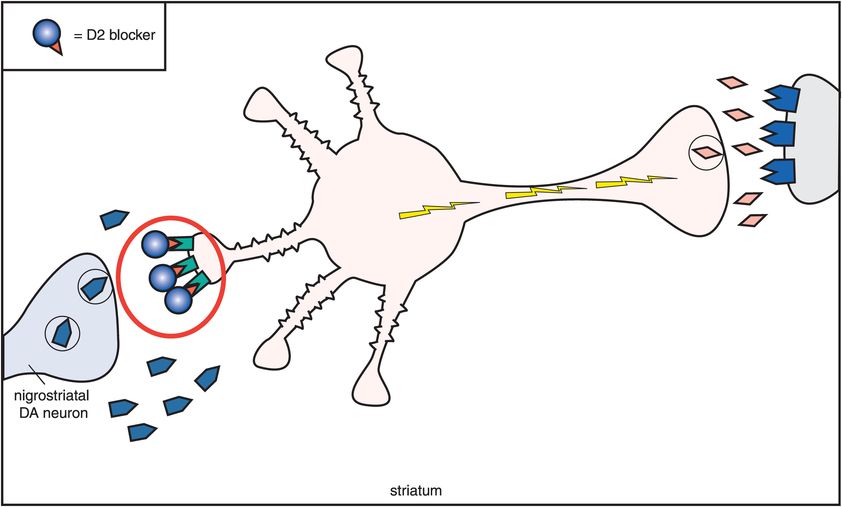
B. Dopamine, acetylcholine, and D2 antagonism. This figure shows what happens to acetylcholine activity when dopamine receptors are blocked. As dopamine normally suppresses acetylcholine activity, removal of dopamine inhibition causes an increase in acetylcholine activity. Thus if dopamine receptors are blocked at the D2 receptors on the cholinergic dendrite on the left, then acetylcholine becomes overly active, with enhanced release of acetylcholine from the cholinergic axon on the right. This is associated with the production of extrapyramidal symptoms (EPS). The pharmacological mechanism of EPS therefore seems to be a relative dopamine deficiency and a relative acetylcholine excess.
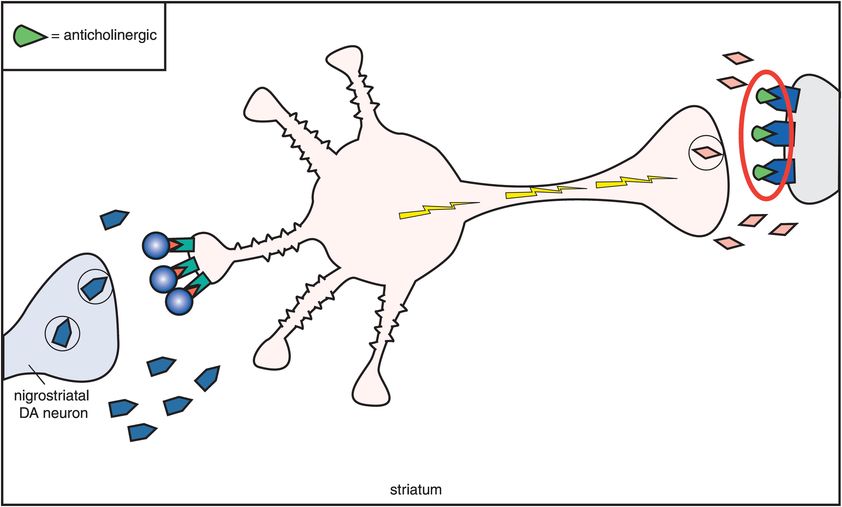
C. D2 antagonism and anticholinergic agents. One compensation for the overactivity that occurs when dopamine receptors are blocked is to block the acetylcholine receptors with an anticholinergic agent (M1 receptors being blocked by an anticholinergic on the far right). Thus, anticholinergics overcome excess acetylcholine activity caused by removal of dopamine inhibition when dopamine receptors are blocked by conventional antipsychotics. This also means that extrapyramidal symptoms (EPS) are reduced.
One compensation for this overactivity of acetylcholine is to block it with an anticholinergic agent (Figure 5-11C). Thus, drugs with anticholinergic actions will diminish the excess acetylcholine activity caused by removal of dopamine inhibition when dopamine receptors are blocked (Figures 5-10 and 5-11C). If anticholinergic properties are present in the same drug with D2 blocking properties, they will tend to mitigate the effects of D2 blockade in the nigrostriatal dopamine pathway. Thus, conventional antipsychotics with potent anticholinergic properties have lower EPS than conventional antipsychotics with weak anticholinergic properties. Furthermore, the effects of D2 blockade in the nigrostriatal system can be mitigated by co-administering an agent with anticholinergic properties. This has led to the common strategy of giving anticholinergic agents along with conventional antipsychotics in order to reduce EPS. Unfortunately, this concomitant use of anticholinergic agents does not lessen the ability of the conventional antipsychotics to cause tardive dyskinesia. It also causes the well-known side effects associated with anticholinergic agents, such as dry mouth, blurred vision, constipation, urinary retention, and cognitive dysfunction (Figure 5-10).
Other pharmacologic properties of conventional antipsychotic drugs
Still other pharmacologic actions are associated with the conventional antipsychotic drugs. These include generally undesired blockade of histamine H1 receptors (Figure 5-9) causing weight gain and drowsiness, as well as blockade of α1-adrenergic receptors causing cardiovascular side effects such as orthostatic hypotension and drowsiness. Conventional antipsychotic agents differ in terms of their ability to block these various receptors represented in Figure 5-9. For example, the popular conventional antipsychotic haloperidol has relatively little anticholinergic or antihistaminic binding activity, whereas the classic conventional antipsychotic chlorpromazine has potent anticholinergic and antihistaminic binding. Because of this, conventional antipsychotics differ somewhat in their side-effect profiles, even if they do not differ overall in their therapeutic profiles. That is, some conventional antipsychotics are more sedating than others, some have more ability to cause cardiovascular side effects than others, some have more ability to cause EPS than others.
A somewhat old-fashioned way to subclassify conventional antipsychotics is “low potency” versus “high potency” (Table 5-1). In general, as the name implies, low-potency agents require higher doses than high-potency agents, but, in addition, low-potency agents tend to have more of the additional properties discussed here than do the so-called high-potency agents: namely, low-potency agents have greater anticholinergic, antihistaminic, and α1 antagonist properties than high-potency agents, and thus are probably more sedating in general. A number of conventional antipsychotics are available in long-acting depot formulations (Table 5-1).
Atypical antipsychotics
What makes an antipsychotic “atypical”?
From a clinical perspective, an “atypical antipsychotic” is defined in part by the “atypical” clinical properties that distinguish such drugs from conventional antipsychotics. That is, atypical antipsychotics have the clinical profile of equal positive symptom antipsychotic actions, but low extrapyramidal symptoms and less hyperprolactinemia compared to conventional antipsychotics. Thus, they are “atypical” from what is expected from a classical, conventional, first-generation antipsychotic. Since almost all of the agents with this atypical profile came after the introduction of clozapine, sometimes the atypical antipsychotics are also called second-generation antipsychotics.
From a pharmacological perspective, the current atypical antipsychotics as a class are defined as serotonin–dopamine antagonists, with simultaneous serotonin 5HT2A receptor antagonism that accompanies D2 antagonism (Figure 5-12). Pharmacologic actions in addition to 5HT2A antagonism that can hypothetically also mediate the atypical antipsychotic clinical profile of low EPS and less hyperprolactinemia with comparable antipsychotic actions include partial agonist actions at 5HT1A receptors and partial agonist actions at D2 receptors. Each of these mechanisms will be discussed here. In order to understand the mechanism of action of atypical antipsychotics and how this differs from conventional antipsychotics, it is necessary to have a somewhat detailed understanding of the neurotransmitter serotonin and its receptors; thus serotonin pharmacology will be discussed in detail throughout this chapter.
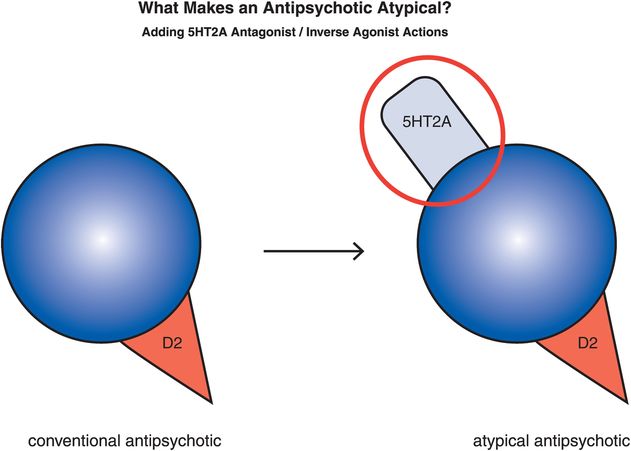
Figure 5-12. Serotonin–dopamine antagonist. The “atypicality” of atypical antipsychotics has often been attributed to the coupling of D2 antagonism with serotonin 5HT2A antagonism. On the right is an icon representing this dual pharmacological action.
Serotonin synthesis and termination of action
Serotonin is also known as 5-hydroxytryptamine and abbreviated as 5HT. Synthesis of 5HT begins with the amino acid tryptophan, which is transported into the brain from the plasma to serve as the 5HT precursor (Figure 5-13). Two synthetic enzymes then convert tryptophan into serotonin: firstly tryptophan hydroxylase (TRY-OH) converts tryptophan into 5-hydroxytryptophan, and then aromatic amino acid decarboxylase (AAADC) converts 5HTP into 5HT (Figure 5-13). After synthesis, 5HT is taken up into synaptic vesicles by a vesicular monoamine transporter (VMAT2) and stored there until it is used during neurotransmission.
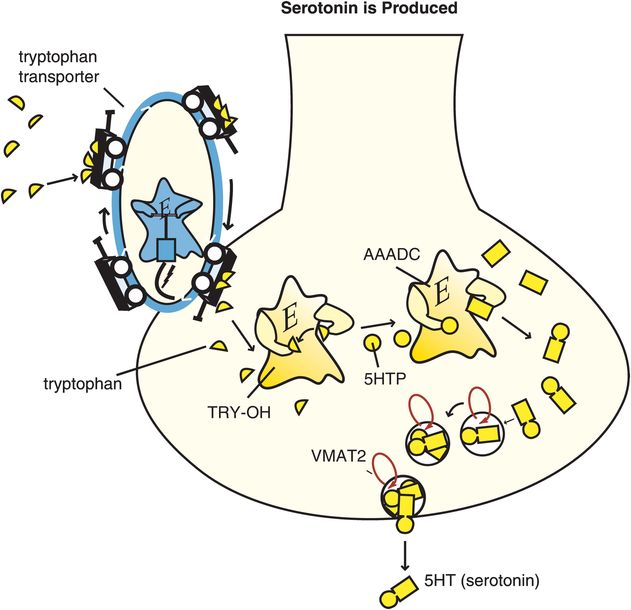
Figure 5-13. Serotonin is produced. Serotonin (5-hydroxytryptamine [5HT]) is produced from enzymes after the amino acid precursor tryptophan is transported into the serotonin neuron. The tryptophan transport pump is distinct from the serotonin transporter. Once transported into the serotonin neuron, tryptophan is converted by the enzyme tryptophan hydroxylase (TRY-OH) into 5-hydroxytryptophan (5HTP), which is then converted into 5HT by the enzyme aromatic amino acid decarboxylase (AAADC). Serotonin is then taken up into synaptic vesicles via the vesicular monoamine transporter (VMAT2), where it stays until released by a neuronal impulse.
5HT action is terminated when it is enzymatically destroyed by monoamine oxidase (MAO), and converted into an inactive metabolite (Figure 5-14). Serotonergic neurons themselves contain MAO-B, which has low affinity for 5HT, so much of 5HT is thought to be enzymatically degraded by MAO-A outside of the neuron once 5HT is released. The 5HT neuron also has a presynaptic transport pump for serotonin called the serotonin transporter (SERT) that is unique for 5HT and that terminates serotonin’s actions by pumping it out of the synapse and back into the presynaptic nerve terminal where it can be re-stored in synaptic vesicles for subsequent use in another neurotransmission (Figure 5-14).
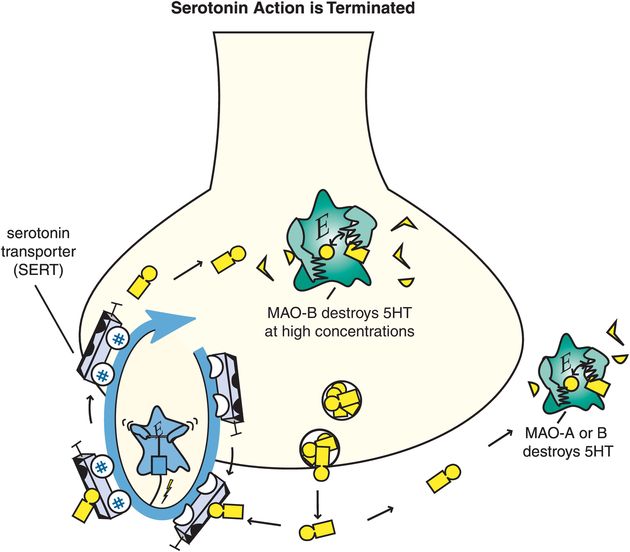
Figure 5-14. Serotonin’s action is terminated. Serotonin (5HT) action is terminated by the enzymes monoamine oxidase A (MAO-A) and MAO-B outside the neuron, and by MAO-B within the neuron when it is present in high concentrations. These enzymes convert serotonin into an inactive metabolite. There is also a presynaptic transport pump selective for serotonin, called the serotonin transporter or SERT, that clears serotonin out of the synapse and back into the presynaptic neuron.
5HT2A receptors
The key to understanding why antipsychotics are atypical is to understand the pharmacology of 5HT2A receptors, and the significance of what happens when they are blocked by atypical antipsychotics. All 5HT2A receptors are postsynaptic, and 5HT2A receptors are located in many brain regions. When they are located on cortical pyramidal neurons, they are excitatory (Figure 5-15A, box 1) and can thus enhance downstream glutamate release (Figure 5-15A, box 2). As discussed in Chapter 4, glutamate regulates downstream dopamine release, so stimulating (Figure 5-15A) or blocking (Figure 5-15B) 5HT2A receptors can therefore also regulate downstream dopamine release. Cortical 5HT1A receptors also regulate downstream dopamine release (Figure 5-15C, discussed below).
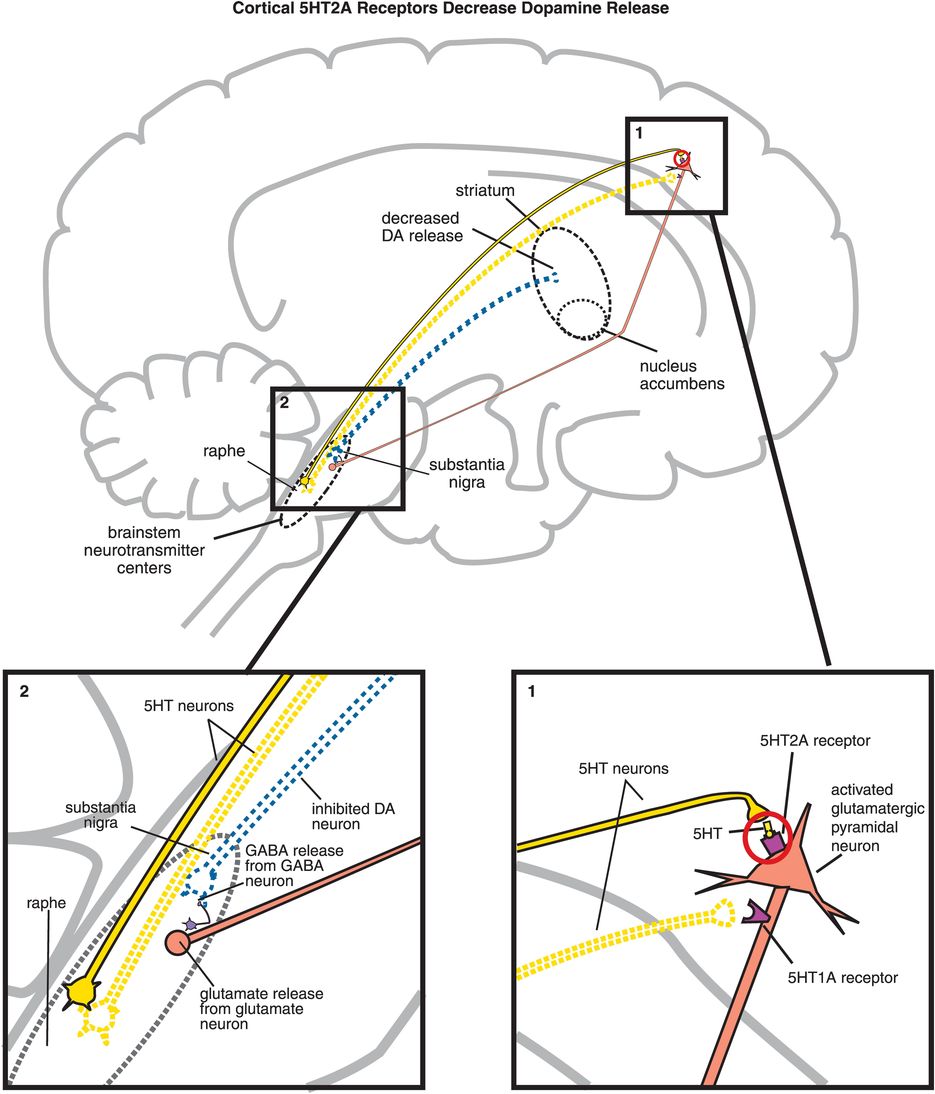
A. Cortical 5HT2A receptors decrease dopamine release. Shown here is the mechanism by which serotonin release in the cortex can lead to decreased dopamine release in the striatum. (1) Serotonin is released in the cortex and binds to 5HT2A receptors on glutamatergic pyramidal neurons, causing activation of those neurons. (2) Activation of glutamatergic pyramidal neurons leads to glutamate release in the brainstem, which in turn stimulates GABA release. GABA binds to dopaminergic neurons projecting from the substantia nigra to the striatum, inhibiting dopamine release (indicated by the dotted outline of the dopaminergic neuron).
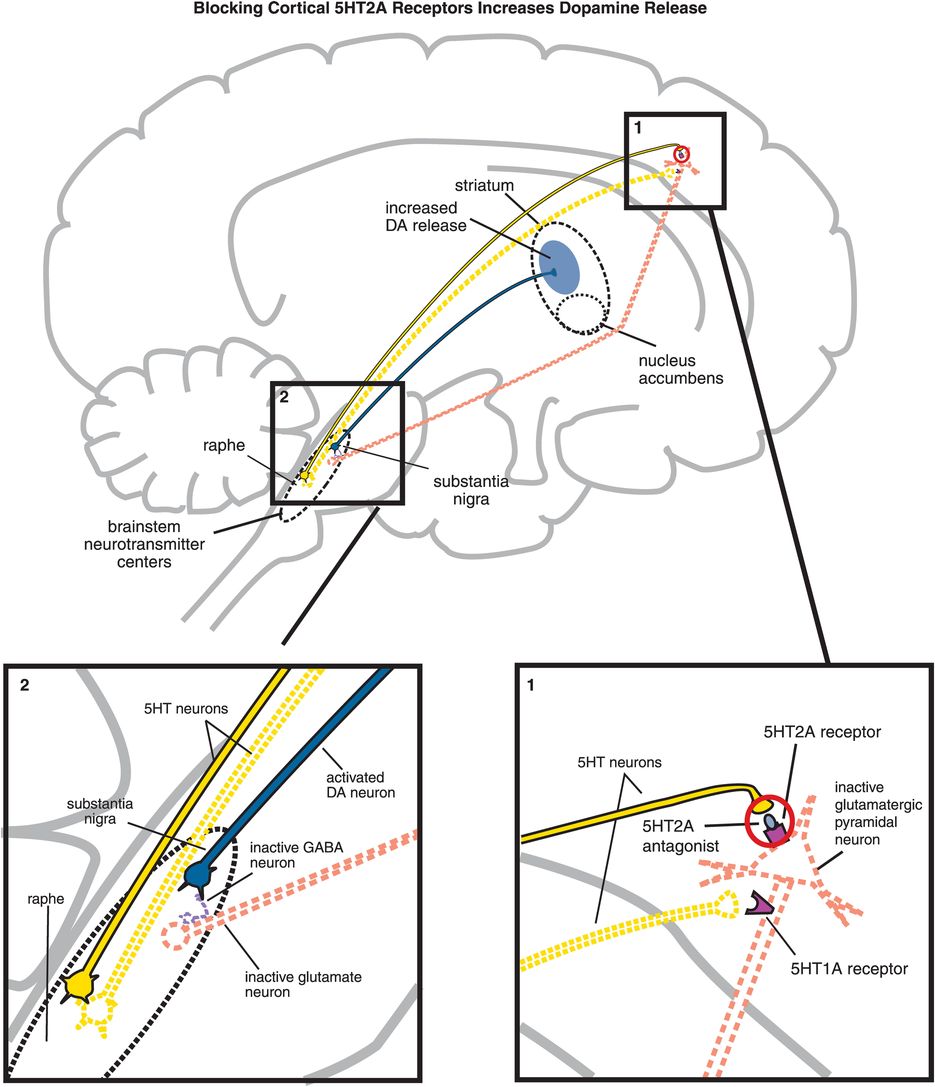
B. Blocking cortical 5HT2A receptors increases dopamine release. (1) If 5HT2A receptors on glutamatergic pyramidal neurons are blocked, then these neurons cannot be activated by serotonin release in the cortex (indicated by the dotted outline of the glutamatergic neuron). (2) If glutamate is not released from glutamatergic pyramidal neurons into the brainstem, then GABA release is not stimulated and in turn cannot inhibit dopamine release from the substantia nigra into the striatum.
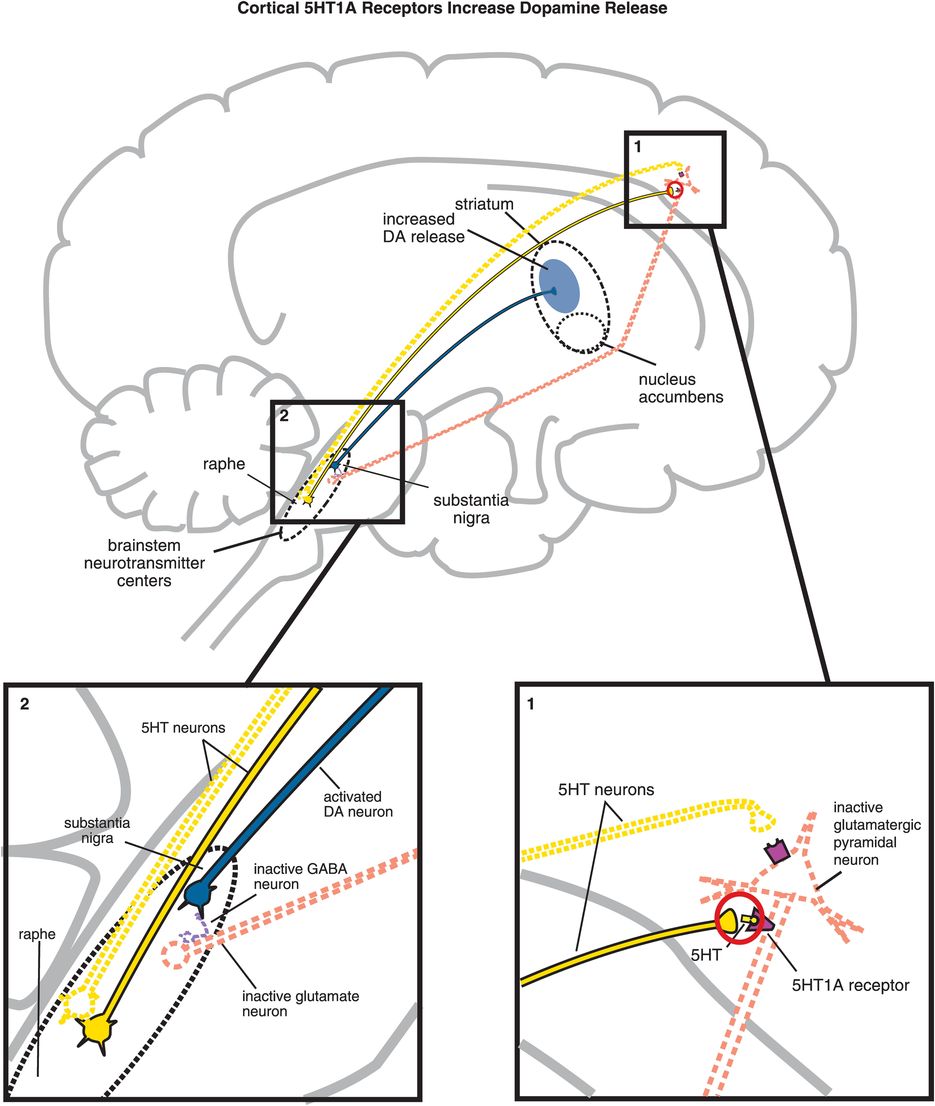
C. Cortical 5HT1A receptor stimulation increases dopamine release. Serotonin projections from the raphe nucleus to the cortex also make axoaxonic connections with glutamatergic pyramidal neurons. (1) Serotonin released at these synapses can bind to 5HT1A receptors, which causes inhibition of the glutamatergic neuron (indicated by the dotted outline of the glutamatergic neuron). (2) If glutamate is not released from glutamatergic pyramidal neurons into the brainstem, then GABA release is not stimulated and in turn cannot inhibit dopamine release from the substantia nigra into the striatum. Thus, cortical 5HT1A receptor stimulation is functionally analogous to cortical 5HT2A receptor blockade, in that both lead to increased dopamine release in the striatum.
5HT2A receptors are brakes on dopamine release in the striatum
5HT2A stimulation of cortical pyramidal neurons by serotonin (Figure 5-15A, box 1) hypothetically blocks downstream dopamine release in the striatum. It does this via stimulation of glutamate release in the brainstem that triggers release of inhibitory GABA there (Figure 5-15A, box 2). Release of dopamine from neurons in the striatum is thus inhibited (Figure 5-15A).
5HT2A antagonism cuts the brake cable
5HT2A antagonism of cortical pyramidal neurons by an atypical antipsychotic interferes with serotonin applying its braking action to dopamine release via 5HT2A receptors (Figure 5-15B, box 1). Thus, 5HT2A antagonism in the cortex hypothetically stimulates downstream dopamine release in the striatum (Figure 5-15B). It does this by reducing glutamate release in the brainstem, which in turn fails to trigger the release of inhibitory GABA at dopamine neurons there (Figure 5-15B, box 2). Release of dopamine from neurons downstream in the striatum is thus disinhibited, which should theoretically mitigate EPS.
5HT2A receptors in other brain areas are also a brake on dopamine release in the striatum
5HT2A receptors theoretically regulate dopamine release from nigrostriatal dopamine neurons by additional mechanisms in additional brain areas. That is, serotonin neurons whose cell bodies are in the midbrain raphe may innervate nigrostriatal dopamine neurons both at the level of the dopamine neuronal cell bodies in the substantia nigra (Figure 5-16A, box 2) and at the dopamine neuronal axon terminals in the striatum (Figure 5-16A, box 1). This innervation may be either via a direct connection between the serotonin neuron and the dopamine neuron, or via an indirect connection with a GABA interneuron. 5HT2A receptor stimulation by serotonin at either end of substantia nigra neurons hypothetically blocks dopamine release in the striatum (Figure 5-16A). On the other hand, 5HT2A receptor antagonism by an atypical antipsychotic at these same sites hypothetically stimulates downstream dopamine release in the striatum (Figure 5-16B). Such release of dopamine in the striatum should mitigate EPS, which is theoretically why antipsychotics with 5HT2A antagonist properties are atypical. 5HT1A receptors also regulate dopamine release in the striatum (Figure 5-16C, discussed below).
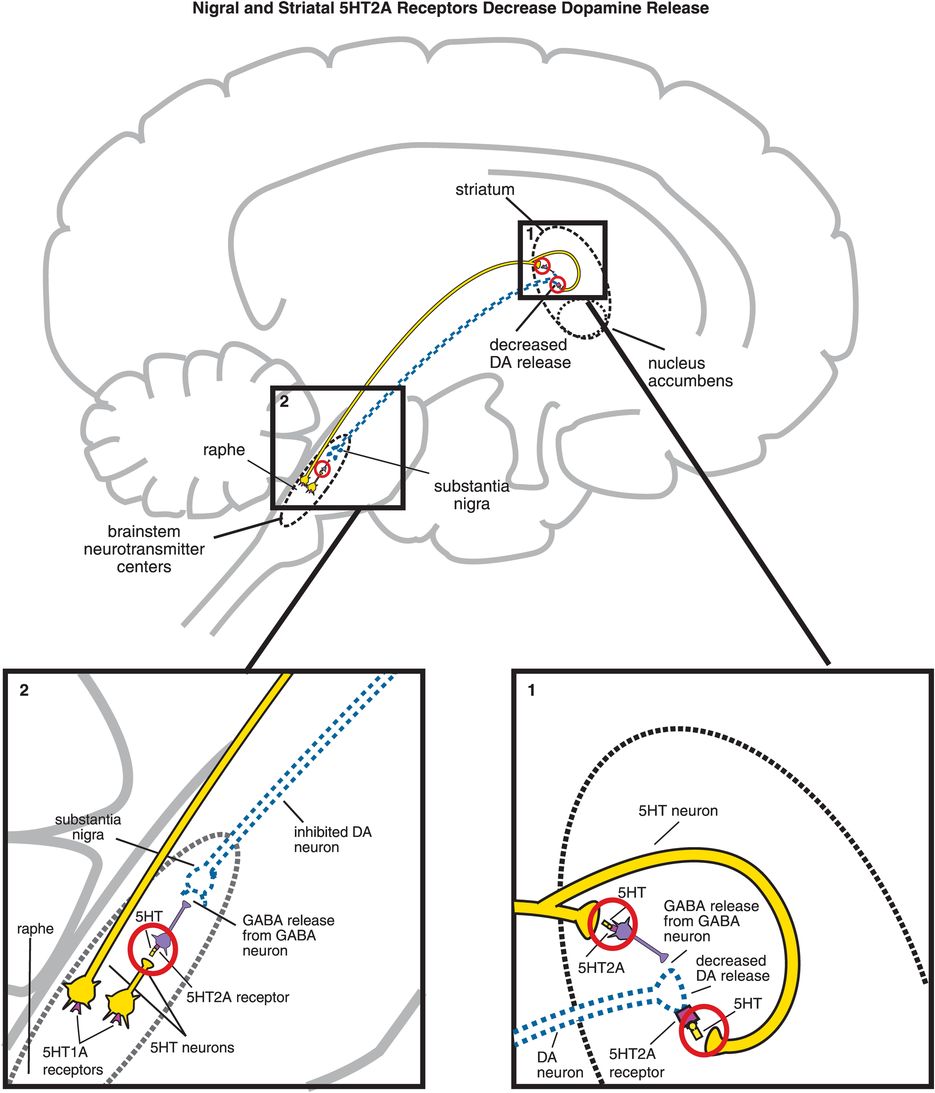
A. Nigral and striatal 5HT2A receptor stimulation decreases dopamine release. (1) In the striatum, serotonergic projections synapse directly with dopaminergic neurons and indirectly via GABAergic neurons. At GABAergic neurons, serotonin binding to 5HT2A receptors disinhibits GABA release, which in turn decreases release of dopamine (indicated by the dotted outline of the dopaminergic neuron). Similarly, when serotonin binds to 5HT2A receptors directly on dopamine neurons, this causes a decrease in dopamine release. (2) Serotonin can also decrease dopamine release in the striatum via 5HT2A binding in the brainstem. That is, serotonin released in the raphe nucleus binds to 5HT2A receptors on GABAergic interneurons. This causes GABA to be released onto dopaminergic neurons in the substantia nigra, thus inhibiting dopamine release into the striatum (indicated by the dotted outline of the dopaminergic neuron).
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


