and FJE Vajda2
(1)
Clinical Neurology and Neuropharmacology, University of Queensland, and Honorary Consultant Neurologist, Royal Brisbane and Women’s Hospital, Brisbane, QLD, Australia
(2)
Department of Medicine and Neurology Director of the Australian Epilepsy and Pregnancy Register, University of Melbourne and Royal Melbourne Hospital, Melbourne, Australia
Abstract
This chapter contains information concerning the dispositions of various antiepileptic drugs that have sometimes been used in pregnancy, in particular oxcarbazepine. The information concerning the effects of pregnancy on the pharmacokinetics of gabapentin, ethosuximide, vigabatrin, zonisamide and various benzodiazepines with antiepileptic properties is relatively scanty, though the available data do not suggest that there are significant departures from the principles set down in Chap. 3 in relation to the anticipated effects of pregnancy on drug disposition.
Some information is available concerning the dispositions during pregnancy of several antiepileptic drugs that are sometimes taken by contemporary pregnant women. Several of these drugs have been marketed for a number of years but their use is diminishing as time passes. Others are relatively new agents whose place in therapeutics is not yet well established. One, ethosuximide , has a long established but limited and specific place in treating a type of epileptic disorder that is not often encountered in pregnant women. The drugs are dealt with individually in this chapter, though in less detail than the more commonly used drugs considered in the previous two chapters. There is little useful information available in the literature concerning the dispositions in pregnancy of a few older antiepileptic drugs that may still be taken by a few pregnant women, e.g. sulthiame and felbamate . At the time of writing, there is virtually no relevant information for certain newer antiepileptic drugs such as lacosamide , perampanel and eslicarbazepine , which are beginning to play increasing roles in the treatment of epilepsy but do not yet appear to have become widely used in pregnancy.
Oxcarbazepine
Rather more information is available concerning the disposition in pregnancy of oxcarbazepine than that for the other drugs that are to be considered in this chapter. Oxcarbazepine has been available for a number of years and seems to have been rather extensively used in some European countries but less widely employed elsewhere. The drug has molecular structural and pharmacological similarities to carbamazepine.
Flesch (2004) has published a detailed account of the clinical pharmacology of the drug.
Chemistry
Oxcarbazepine (10,11-dihydro-10-oxo-5H-dibenz[b,f]azepine-5-carboxamide) is a white, poorly water-soluble material (molecular weight 252.3, pKa value 10.7), which is supplied for therapeutic purposes in 150, 300 and 600 mg solid dosage forms and in a 60 mg per ml suspension. Its molecular structural formula is shown in Fig. 4.6 in relation to that of carbamazepine .
Pharmacodynamics
In clinical use, oxcarbazepine in effect functions mainly as a prodrug for its pharmacologically active 10-hydroxy metabolite (whose name is often abbreviated to MHD, i.e. monohydroxy derivative, whose molecular formula also appears in Fig. 4.6). The mechanism of action of this substance is very similar to that of carbamazepine , viz. blocking voltage- and frequency-dependent Na+ ion channels in neural cell membranes. The monohydroxy derivative of oxcarbazepine and carbamazepine itself both act at Ca2+ channels , but the actions of the two rather structurally similar molecules occur at different types of Ca2+ channel (Schmidt and Elger 2004). Oxcarbazepine is useful in controlling the seizures of the focal epilepsies and the generalised tonic–clonic convulsions of genetic generalised epilepsy , but the drug is not useful for preventing absence or myoclonic seizures .
Pharmacokinetics
The pharmacokinetics of oxcarbazepine is linear under the conditions that apply in human therapeutic use.
Absorption
Orally administered oxcarbazepine appears to be fully bioavailable.
Distribution
The apparent volume distribution of the drug was found to be 49 L, suggesting that it is probably distributed throughout body water. Some two-thirds of the oxcarbazepine in the circulation and 40 % of its monohydroxy metabolite are bound to plasma proteins.
Elimination
The plasma elimination half-life of oxcarbazepine is in the range 1.3–2.3 h, whereas the half-life of the monohydroxy derivative is 9.3 ± 1.8 h. The clearance value of the monohydroxy derivative is 91–122 ml per minute [5.46–7.32 L/h].
Excretion Unchanged
Less than 1 % of an oxcarbazepine dose appears in urine as the unmetabolised parent substance.
Metabolism
Oxcarbazepine is converted to its hydroxy metabolite by arylketone reductase enzymes in the cytosol; the majority of this metabolite is then conjugated with glucuronic acid before excretion in the urine. The monohydroxy derivative exist in two stereoisomeric forms, the [S]-enantiomer predominating. Both enantiomers are pharmacologically active. Part of the monohydroxy metabolite is converted to a biologically inactive dihydrodiol derivative that is subsequently excreted in urine mainly as a glucuronide conjugate. This same dihydrodiol metabolite is formed from carbamazepine via the epoxide hydrolase pathway that is involved in carbamazepine’s biotransformation (see Fig. 4.6).
Clinical Pharmacokinetics
In human therapeutic use, plasma concentrations of the monohydroxy derivative of oxcarbazepine increase in proportion to the oxcarbazepine dose. Patsalos et al. (2008) proposed a plasma therapeutic range of 3–35 mg/L for this monohydroxy derivative .
Interactions
Neither oxcarbazepine nor its monohydroxy derivative induces the CYP450 enzyme system, though both substances inhibit the isoenzyme CYP2C19 . The monohydroxy derivative does not alter circulating concentrations of phenytoin , carbamazepine or valproate . However, in the case of co-administered carbamazepine, it causes raised circulating concentrations of carbamazepine-10,11-epoxide . Oxcarbazepine administration may lower plasma concentrations of lamotrigine .
Co-administered phenytoin , carbamazepine and valproate will reduce circulating concentrations of oxcarbazepine’s monohydroxy derivative .
Adverse Effects
Oxcarbazepine is less likely than carbamazepine to produce skin rashes and tends to cause less sedation at doses which produce similar degrees of antiepileptic effect. Unfortunately, the drug has a greater tendency to produce hyponatraemia , particularly early in the course of its use. This hyponatraemia may sometimes be severe enough to compromise seizure control.
Pregnancy
The Mother
In five women, Mazzucchelli et al. (2006) found that, during pregnancy, plasma concentrations of the [S]-isomer of the monohydroxy derivative of oxcarbazepine fell relative to the oxcarbazepine dose. The plasma concentration of oxcarbazepine itself also fell. Christensen et al. (2006) followed the relationship between plasma monohydroxy derivative concentrations and the oxcarbazepine dose throughout nine pregnancies that occurred in seven women. They found that the relationship between the plasma metabolite concentrations and the drug dose decreased to 72 % of its pre-pregnancy value in the first trimester, to 74 % in the second and to 64 % in the third. They suggested that the increased clearance of the metabolite probably depended on increased formation and renal excretion of its glucuronide conjugate, though they did not prove that this was the case. Subsequently Petrenaite et al. (2009) carried out a rather similar study with a similar result, the ratio between plasma metabolite concentration and the drug dose falling by 26.2 % in the first trimester, by 36.5 % in the second and by 38.2 % in the third. In the following year, Wegner et al. (2010) also noted an increased clearance of the metabolite in two pregnancies. There thus seems consistent evidence that the drug and its major biologically active metabolite undergo increased clearances during pregnancy, though the details of the responsible mechanism have not been established.
The Foetus
In ex vivo perfusion experiments, Pienimäki et al. (1997) showed that oxcarbazepine was converted to its monohydroxy derivative in the human placenta . The monohydroxy derivative was not metabolised further in that organ. It appears that the drug probably equilibrates crosses the placenta, at least in late pregnancy. Myllynen et al. (2001) found that concentrations of oxcarbazepine and its monohydroxy and dihydrodiol derivatives were similar in maternal and umbilical cord serum shortly after birth. Measurable quantities of oxcarbazepine, its monohydroxy derivative and its dihydrodiol derivative were present in neonatal plasma shortly after birth.
Breast Milk
Breast milk concentrations of drug and its main metabolite are about half those in simultaneously collected maternal plasma. Davanzo et al. (2013) also cited the same 50 % figure.
The Neonate
It has not been possible to trace information about the elimination of the drug or its biologically active metabolite in the neonate.
Gabapentin
Gabapentin was synthesised in an attempt to produce a molecule with structural similarities to the inhibitory amino acid neurotransmitter γ-aminobutyrate (GABA ) but with an increased lipophilicity that might permit it to cross the blood–brain barrier and in that organ mimic the effects of GABA . The drug has been marketed for some 20 years and has an established role as a moderately effective antiepileptic agent.
Chemistry
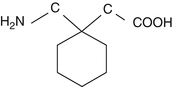
Gabapentin (1-aminomethyl-cyclohexyl-acetic acid) is a water-soluble white crystalline substance (molecular weight 171.34, pKa values 3.68 and 10.70). It is marketed in 100, 300, 400, 600 and 800 mg solid dosage forms and in a solution in a concentration of 50 mg per ml.
Pharmacodynamics
Despite the concept which led to the development of gabapentin , viz. the idea that it would reproduce the inhibitory neurotransmitter effects of GABA within the central nervous system, there is little convincing evidence that the drug acts through this mechanism. Instead, the drug appears to bind specifically to a protein which forms part of voltage-gated Ca2+ channels (Striano and Striano 2008), though exactly how this binding translates into an antiepileptic effect is not yet fully explained.
Clinically, the drug demonstrates efficacy against focal (partial) seizures . Its role in the treatment of genetic generalised tonic–clonic seizures is not well established and it appears ineffective in managing absence seizures . It has also found a use in treating neuropathic pain .
Pharmacokinetics
Absorption
Orally administered gabapentin is absorbed from the alimentary tract through the saturable active transport mechanism in the gut wall that is responsible for the uptake of L-amino acids. Because of its potentially saturable absorption, the oral bioavailability of the drug, which is around 60 % for a 300 mg dose, lessens with the increasing size of individual oral drug doses.
Distribution
Gabapentin does not bind to plasma proteins, has an apparent volume of distribution (approximately 0.9 L per kilogram) that is a little greater than the volume of total body water and enters the brain by active transport mediated by the L-amino acid uptake mechanism in the blood–brain barrier. The gabapentin concentration in CSF can be anywhere between 7 % and 35 % of its simultaneous concentration in plasma.
Elimination
The half-life of gabapentin is 5–7 h and the plasma clearance around 0.9 L per kilogram per hour. The half-life is inversely related to the creatinine clearance.
Excretion Unchanged
No metabolite of gabapentin has been found in human urine. Apparently the entire absorbed dose of the drug is excreted intact.
Metabolism
In humans, as mentioned immediately above, gabapentin forms no known biotransformation products.
Clinical Pharmacokinetics
Steady-state conditions should apply approximately 2 days after a gabapentin dosage change. The clearance of the drug tends to be inversely related to the efficiency of renal function.
Patsalos et al. (2008) proposed that the therapeutic range of plasma concentrations for the drug was between 2 and 20 mg/L.
Interactions
Since gabapentin is not metabolised in the human body, it does not interact with co-administered drugs, including antiepileptic agents, which are cleared from the human body by biotransformation. It also does not interact with the steroidal sex hormones contained in combined oral contraceptive pills (Eldon et al. 1998). Virtually no interactions involving the drug are known, though antacids containing aluminium or magnesium hydroxides may interfere with its absorption from the alimentary tract.
Adverse Effects
The main adverse effects of the drug include drowsiness and sometimes dizziness, unsteadiness, headache, and double vision . Gabapentin seems to have very few serious idiosyncratic unwanted effects.
Pregnancy
Relatively little information is available regarding the disposition of gabapentin in the mother and her foetus.
The Mother
No relevant data have been found in the literature.
The Foetus
At birth, Öhman et al. (2005) found that, in five mother–infant pairs, the average umbilical cord vein gabapentin concentration was 1.7 times (range 1.3–2.1) that in maternal plasma.
Breast Milk
Öhman et al. (2005) also found that, between 2 weeks and 3 months after childbirth, gabapentin concentrations were similar in maternal plasma and in breast milk . This is not surprising, because the drug is not bound to plasma proteins and probably simply equilibrates between the two fluids. Davanzo et al. (2013) stated that the milk concentration could be between 70 and 130 % of that in maternal plasma.
The Neonate
According to Öhman et al. (2005), in the breastfed baby, the plasma gabapentin concentration was approximately 12 % of that in the mother, with the drug’s elimination half-life being 14 h.
Vigabatrin
Vigabatrin was developed with the strategic intention of raising brain concentrations of the inhibitory neurotransmitter GABA by inactivating the enzyme responsible for catalysing its catabolism, viz. GABA transaminase . For a time the drug seemed to offer very useful therapeutic benefits in treating focal epilepsies . However, the subsequent recognition that vigabatrin could be responsible for serious adverse effects involving vision has considerably limited the drug’s use.
Chemistry
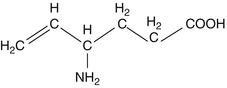
Vigabatrin (γ-vinyl-GABA) is a white, water-soluble material (molecular weight 129.16) which is supplied commercially as a mix of equal parts of the [S]- and [R]-stereoisomers, though only the former is biologically active. It is marketed in a 500 mg solid dosage form and as a powder.
Pharmacodynamics
Vigabatrin is an irreversible inhibitor of the enzyme GABA transaminase . After a dose, the drug’s action persists, irrespective of the presence of intact vigabatrin in biological fluids, until new transaminase is synthesised. The drug was being used with success in treating focal epilepsies and some varieties of genetic generalised epilepsies , until the frequency of the visual problems associated with the drug’s use was recognised. At the present time, vigabatrin’s use is largely confined to the treatment of myoclonic epilepsies in the very young, in particular infantile spasms .
Pharmacokinetics
Absorption
Based on urinary excretion data, the oral bioavailability of the [S]-enantiomer of vigabatrin appears to be at least 50 % and that of the biologically inactive [R]-enantiomer 65 % or greater.
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


