and FJE Vajda2
(1)
Clinical Neurology and Neuropharmacology, University of Queensland, and Honorary Consultant Neurologist, Royal Brisbane and Women’s Hospital, Brisbane, QLD, Australia
(2)
Department of Medicine and Neurology Director of the Australian Epilepsy and Pregnancy Register, University of Melbourne and Royal Melbourne Hospital, Melbourne, Australia
Abstract
The progressive physiological changes that occur in the female body during pregnancy, and their reversal in the weeks after the delivery of the foetus and placenta at the time of childbirth, have consequences for the body’s handling of drugs. The physiological changes have little effect on the absorption of the orally administered drugs, but the expanded extracellular fluid volume of pregnancy and the increasing bulk of the uterus and its contents have a diluting effect on circulating drug concentrations. Also, in later pregnancy, plasma protein concentrations decrease, resulting in a relative increase in plasma-unbound drug concentrations relative to total drug concentrations. Overall, the magnitude of these effects is small relative to the effects of pregnancy on drug elimination. Increased glomerular filtration during pregnancy increases the excretion of antiepileptic drugs that are cleared from the body predominantly as unchanged molecules, while the increasing circulating steroidal sex hormone concentrations of pregnancy induce formation of the liver enzymes that metabolise antiepileptic drugs that are cleared from the body by biotransformation. The overall result of these two processes is for circulating concentrations of antiepileptic drugs to fall relative to drug dose during pregnancy, potentially compromising the control of the seizure disorders for which the drugs have been prescribed.
After childbirth, antiepileptic drugs seem to enter maternal milk by a process of passive transfer along concentration gradients, with factors such as the fat and protein content of the milk affecting the drugs’ concentrations in that fluid. These concentrations are generally lower than those simultaneously present in maternal plasma.
The understanding of the way the human body handles antiepileptic drugs has depended to a considerable extent on the ability to measure the concentrations of these drugs that are present in the body during therapeutic use. Despite at least one earlier attempt, reasonably satisfactory assays that permitted these measurements did not become available until 1956. Then Dill et al. (1956) devised an assay for measuring phenytoin and Plaa and Hine (1956) one for measuring both phenytoin and phenobarbitone simultaneously. These particular methods have since been superseded by numerous more convenient, more specific and more sensitive techniques. Once the ability to undertake the measurements existed, interested clinicians began to employ them to help guide the treatment of epilepsy, while the more pharmacologically minded began to utilise the drug concentration data that became available to enhance the understanding of the dispositions of antiepileptic drugs in the human body.
By the 1970s, enough knowledge was available to permit the publication of two collections of papers describing the measurement techniques and some of their applications in clinical practice (Meijer et al. 1973; Pippenger et al. 1978). Around this time, the first reports appeared of measurements of the behaviour of the concentrations of antiepileptic drugs in the serum or plasma of women during the course of pregnancy (Dam et al. 1976; Lander et al. 1977). The drugs that were then measured were phenytoin and phenobarbitone , at that time the most commonly used antiepileptic agents. Both sets of investigators obtained similar findings, ones that were perhaps a little unanticipated in view of the comparatively stable concentration values relative to drug dose that were present over considerable periods of time in individual women while they were not pregnant. The Danish workers showed that, in all 23 women taking phenytoin (with 14 also taking phenobarbitone), plasma phenytoin concentrations relative to drug dose fell appreciably as pregnancy progressed. The plasma ratio of phenobarbitone concentration to dose also fell, though not so much. The changes could begin as early as the 6th week of pregnancy. Postnatally, the concentration to dose ratios for the drugs tended to return progressively to their pre-pregnancy values (the plasma concentration to dose ratio is the reciprocal of the pharmacokinetic parameter, the steady-state clearance , expressed as volume per unit of time). The Australian workers also showed that plasma phenytoin concentrations fell progressively relative to drug dose as pregnancy progressed (in 9 of 10 women taking the drug), with the concentration to dose ratios returning to their pre-pregnancy values in the 7 of the 9 women whose drug concentrations had been followed into the puerperium (Fig. 3.1). In 5 women taking phenobarbitone, or a structurally related barbiturate molecule that is metabolised to it (methylphenobarbitone or primidone ), the plasma phenobarbitone concentration to dose ratios also fell during pregnancy.
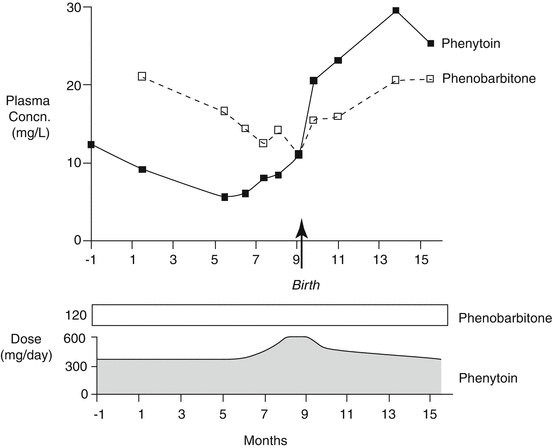
Fig. 3.1
The courses of plasma phenytoin and phenobarbitone concentrations during pregnancy and the puerperium in one woman who took a constant dose of phenobarbitone throughout but whose phenytoin doses were adjusted to try to maintain its plasma concentration in the range 10–20 mg/L (Redrawn from the data of Lander et al. (1977))
The Danish investigators suggested that women developed an increased capacity to metabolise phenytoin during pregnancy. The Australian workers proposed a similar interpretation but in addition raised the possibility that the increased maternal body volume resulting from the presence of the placental –foetal unit together with the possible additional drug-metabolising capacity of that unit, and the known effect of prescribed folic acid intake in reducing plasma phenytoin concentrations, might also contribute to the phenomenon. Because the concentration to dose ratios did not return to pre-pregnancy values immediately after the foetus and placenta became abstracted from the maternal body at the time of childbirth, they favoured the view that changes in the mother rather than the contributions of the foetus and placenta were the main factor involved in the increased clearances of the two drugs during pregnancy.
By the 1970s, the centuries-old notion that drugs acted by means of some mysterious inherent property had almost completely given way to the realisation that the body handled exogenous chemicals, including drugs, through the same physicochemical mechanisms that it employed in dealing with endogenous molecules. The similarities of the findings of the two papers referred to above and the subsequently publications of these two groups with additional case material (Mygind et al. 1976; Eadie et al. 1977) made it likely that both groups had described a genuine phenomenon related to human pregnancy and that the explanation of its mechanism would lie in knowledge of the changes that occur in a woman’s body during and after pregnancy. From this knowledge, principles might be derived that would be applicable more generally to drugs taken during pregnancy.
Body Changes During Pregnancy
Without going into the physiology of pregnancy in more than superficial detail, fertilisation of the ovum followed by implantation of the embryo in the endometrium initiates a series of changes in a woman’s body. The forming trophoblast begins to secrete chorionic gonadotropin, while the corpus luteum does not regress as it does in the latter stage of the normal menstrual cycle . Instead, the corpus continues to secrete progesterone in increasing amounts, while ovarian secretion of oestrogens continues. The resulting raised circulating steroidal sex hormonal levels begin to produce various tissue changes in the maternal body. After about 10 weeks of pregnancy, the corpus luteum secretion of progesterone diminishes. By this time the placenta has largely taken over the secretion of this hormone and also that of oestrogens. The circulating levels of these steroidal hormones continue to rise till near term. The process of childbirth relatively abruptly removes the foetus and placenta, with their associated oestrogen- and progesterone-secreting capacities, from the overall maternal–foetal complex. After childbirth, the maternal pituitary secretion of prolactin increases, initiating lactation. This series of progressive steroidal sex hormonal events during pregnancy produces various changes in the anatomy, physiology and biochemistry of the female body. Those changes likely to be relevant to the body’s handling of antiepileptic drugs are mentioned below.
Changes in Physiology
By late pregnancy (Heidemann and McClure 2003), the maternal plasma volume will have increased by about 45 % over its pre-pregnancy value, because rising circulating concentrations of progesterone and oestrogen cause the increased secretion of renin and aldosterone . These latter increases produce sodium retention and consequent expansion of the plasma volume . The circulating red cell mass increases by about 20 % during pregnancy so that plasma haemoglobin concentrations fall because of the disproportionately greater expansion in plasma water volume. By two weeks after childbirth, the maternal plasma volume will normally have returned to its pre-pregnancy value. The increased circulating concentrations of oestrogen and progesterone during pregnancy lead to vasodilatation. A hyperkinetic circulatory state develops, with a 50 % increase in cardiac output being present by later pregnancy. The increased cardiac output results in increased regional plasma flow and a raised renal glomerular filtration rate . Maternal tissue growth occurs, particularly involving the breasts and uterus. During pregnancy, no consistent change appears to occur in gastric emptying time or in other measures of alimentary tract motility.
Changes in Biochemistry
During pregnancy there is an overall increase in energy production that is required to sustain the growth of maternal and foetal tissues. Pancreatic insulin secretion increases, but a degree of tissue insulin resistance develops. The renal tubules become less able to resorb glucose from tubular urine. There are changes that involve mainly, but are not necessarily restricted to, the maternal liver. These changes influence the extent of the body’s capacity to synthesise various proteins. Circulating levels of enzymes such as γ-glutamyl transpeptidase, alanine transaminase, aspartate transaminase and lactate dehydrogenase tend to rise in pregnancy, though not to concentrations that would be regarded as pathological in other circumstances. By late pregnancy, overall liver protein synthesis has decreased by up to 25 %, resulting in lowered plasma albumin and α1-acid glycoprotein levels. What is particularly pertinent to the foetal–maternal unit’s handling of many drugs is that changes occur in the synthesis of various enzymes that are normally responsible for the inactivation and elimination of certain endogenous molecules and also that of various xenobiotics (including most of the antiepileptic drugs).
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


