Figure 7-1. Response. When treatment of depression results in at least 50% improvement in symptoms, it is called a response. Such patients are better but not well. Previously, this was considered the goal of depression treatment.
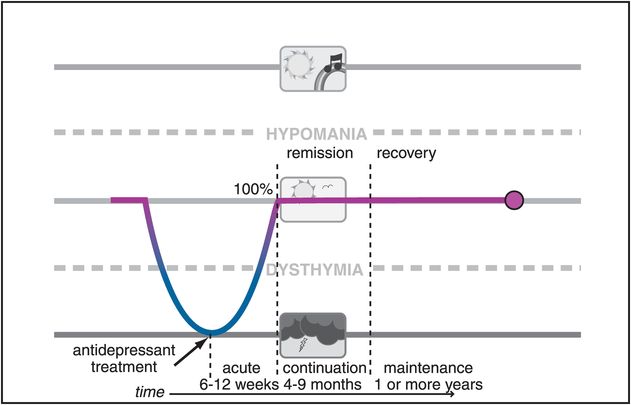
Figure 7-2. Remission. When treatment of depression results in removal of essentially all symptoms, it is called remission for the first several months and then recovery if it is sustained for longer than 6 months. Such patients are not just better – they are well. However, they are not cured, since depression can still recur. Remission and recovery are now the goals when treating patients with depression.
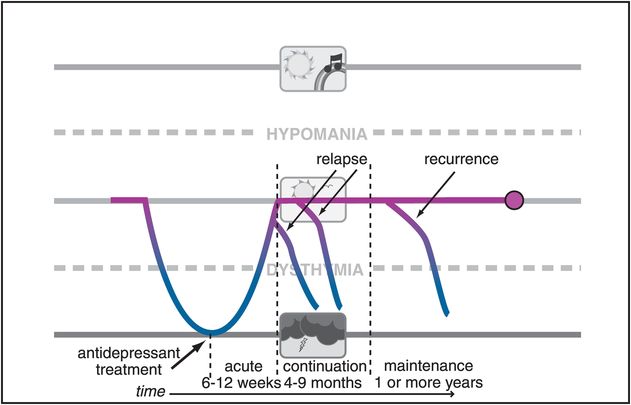
Figure 7-3. Relapse and recurrence. When depression returns before there is a full remission of symptoms or within the first several months following remission of symptoms, it is called a relapse. When depression returns after a patient has recovered, it is called a recurrence.
Do antidepressants work anymore in clinical trials?
Although remission (Figure 7-2) without relapse or recurrence (Figure 7-3) is the widely accepted goal of antidepressant treatment, it is becoming more and more difficult to prove that antidepressants – even well-established antidepressants – work any better than placebo in clinical trials. This is generally due to the fact that in modern clinical trials, the placebo effect has inflated so much over recent decades that placebo now seems to work as well as antidepressants in some trials and nearly as well as antidepressants in other trials. Why this is occurring is the subject of vigorous debate. Some experts propose that it is due to problems conducting clinical ratings in a clinical trial setting that is now unlike a clinical practice setting since patients are seen weekly, often for hours, whether they receive an antidepressant or not; other experts point out that subjects in trials may really be “symptomatic volunteers” who are less ill and less complicated than “real” patients. Critics of psychiatry and psychopharmacology proclaim from clinical trial evidence that antidepressants don’t even work and that their side effects and costs do not justify their use at all. This phenomenon of shrinking and erratic efficacy of long-established antidepressants as well as new antidepressants in clinical trials has also caused the pharmaceutical industry to increasingly abandon the development of new antidepressants. Even patients seem to be affected by this debate, perhaps losing their confidence in the efficacy of antidepressants, since up to a third of patients in a real clinical practice setting never fill their first antidepressant prescription, and for those who do, perhaps less than half get a second month of treatment and maybe less than a quarter get an adequate trial of 3 months or longer. One thing is for sure about antidepressants, and that is that they don’t work if you don’t take them. Thus, the clinical effectiveness of antidepressants in clinical practice settings is reduced by this failure of “persistency” of treatment for a long enough period of time to give the drug a chance to work.
Whatever the cause of this controversy over the efficacy of antidepressants in clinical trials, one only needs to spend a short time in a clinical practice setting to be convinced that antidepressants are powerful therapeutic agents in many patients. Nevertheless, there have been some useful consequences of the debate on the efficacy of antidepressants, such as re-energizing the integration of psychotherapies with antidepressants, searching for new non-medication neurostimulation therapeutics, and studying the combination of currently available antidepressants in order to gain better outcomes, all of which will be discussed in this chapter.
How well do antidepressants work in the real world?
“Real world” trials of antidepressants tested in clinical practice settings that include patients normally excluded from marketing trials, such as the STAR*D trial of antidepressants (Sequenced Treatment Alternatives to Relieve Depression), have recently provided sobering results. Only a third of such patients remit on their first antidepressant treatment, and even after a year of treatment with a sequence of four different antidepressants given for 12 weeks each, only about two-thirds of depressed patients achieve remission of their symptoms (Figure 7-4).
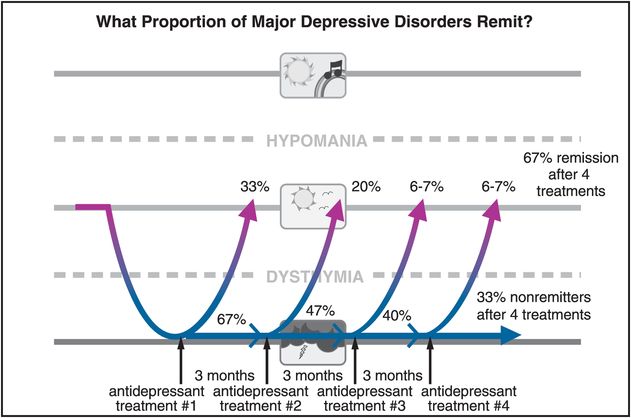
Figure 7-4. Remission rates in MDD. Approximately one-third of depressed patients will remit during treatment with any antidepressant initially. Unfortunately, for those who fail to remit, the likelihood of remission with another antidepressant monotherapy goes down with each successive trial. Thus, after a year of treatment with four sequential antidepressants taken for 12 weeks each, only two-thirds of patients will have achieved remission.
What are the most common symptoms that persist after antidepressant treatment, causing this disorder not to go into remission? The answer is shown in Figure 7-5, and the symptoms include insomnia, fatigue, multiple painful physical complaints (even though these are not part of the formal diagnostic criteria for depression), as well as problems concentrating, and lack of interest or motivation. Antidepressants appear to work fairly well in improving depressed mood, suicidal ideation, and psychomotor retardation (Figure 7-5).
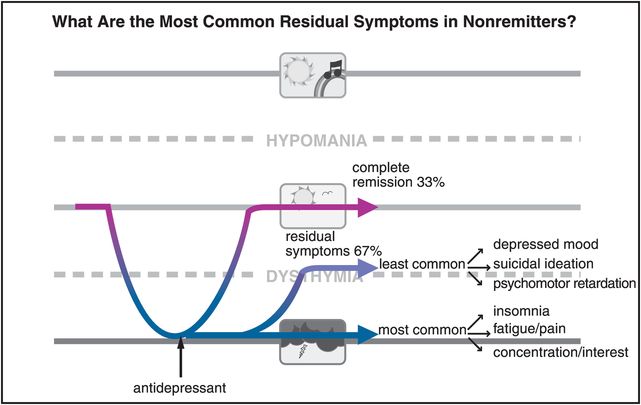
Figure 7-5. Common residual symptoms. In patients who do not achieve remission, the most common residual symptoms are insomnia, fatigue, painful physical complaints, problems concentrating, and lack of interest. The least common residual symptoms are depressed mood, suicidal ideation, and psychomotor retardation.
Why should we care whether a patient is in remission from major depression or has just a few persistent symptoms? The answer can be found in Figure 7-6, which shows both good news and bad news about antidepressant treatment over the long run. The good news is that if an antidepressant gets your patient into remission, that patient has a significantly lower relapse rate. The bad news is that there are still very frequent relapses in the remitters, and these relapse rates get worse the more treatments the patient needs to take in order to get into remission (Figure 7-6).
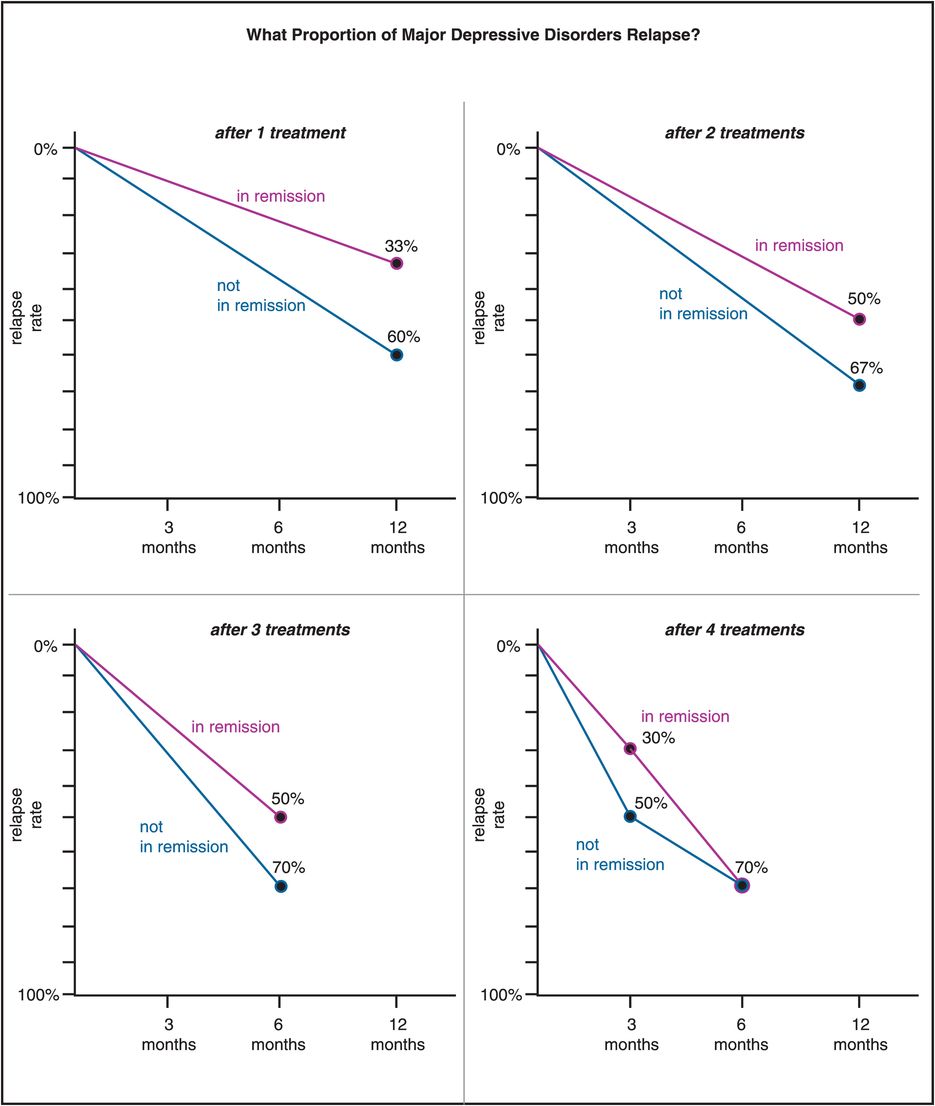
Figure 7-6. Relapse rates. The rate of relapse of major depression is significantly less for patients who achieve remission. However, there is still risk of relapse even in remitters, and the likelihood increases with the number of treatments it takes to get the patient to remit. Thus the relapse rate for patients who do not remit ranges from 60% at 12 months after one treatment to 70% at 6 months after four treatments; but for those who do remit, it ranges from only 33% at 12 months after one treatment all the way to 70% at 6 months after four treatments. In other words, the protective nature of remission virtually disappears once it takes four treatments to achieve remission.
Data like these have galvanized researchers and clinicians alike to treat patients to the point of remission of all symptoms whenever possible, and to try to intervene as early as possible in this illness of major depression, not only to be merciful in trying to relieve current suffering from depressive symptoms, but also because of the possibility that aggressive treatment may prevent disease progression. The concept of disease progression in major depression is controversial, unproven, and provocative, but makes a good deal of sense intuitively for many clinicians and investigators (Figure 6-23). The idea is that chronicity of major depression, development of treatment resistance, and likelihood of relapse could all be reduced, with a better overall outcome, with aggressive treatment of major depressive episodes that leads to remission of all symptoms, thus potentially modifiying the course of this illness. This may pose an especially difficult challenge for treatment of younger patients, where risks versus benefits of antidepressants are currently debated (Figure 7-7).
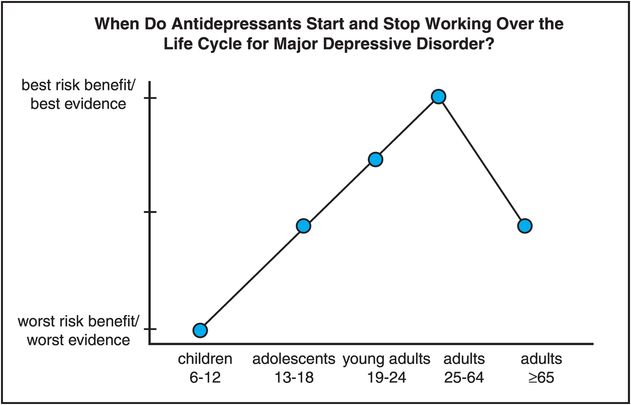
Figure 7-7. Antidepressants over the life cycle. The efficacy, tolerability, and safety of antidepressants have been studied mostly in individuals between the ages of 25 and 64. Existing data across all age groups suggest that the risk–benefit ratio is most favorable for adults between the ages of 25 and 64 and somewhat less so for adults between the ages of 19 and 24, due to a possibly increased risk of suicidality in younger adults. Limited data in children and adolescents also suggest increased risk of suicidality; this, coupled with a lack of data demonstrating clear antidepressant efficacy, gives children between the ages of 6 and 12 the worst risk–benefit ratio, with adolescents intermediate between young adults and children. Elderly patients, 65 years of age and older, may not respond as well or as quickly to antidepressants as other adults and may also experience more side effects than younger adults.
Antidepressants over the life cycle
Adults between the ages of 25 and 64 might have the best chance of getting a good response and with the best tolerability to an antidepressant (Figure 7-7). Adults aged 65 or older may not respond as quickly or as robustly to antidepressants, especially if their first episode starts at this age, and especially when their presenting symptoms are lack of interest and cognitive dysfunction rather than depressed mood, but do not have increased suicidality from taking an antidepressant. At the other end of the adult age range, those younger than 25 may benefit from antidepressant efficacy but with a slightly but statistically greater risk of suicidality (but not completed suicide) (Figure 7-7). Age is thus an important consideration for whether, when, and how to treat a patient with antidepressants throughout the life cycle, and with what potential risk versus benefit.
Antidepressant classes
Blocking monoamine transporters
Classic antidepressant action is to block one or more of the transporters for serotonin, norepinephrine, and/or dopamine. This pharmacologic action is entirely consistent with the monoamine hypothesis of depression, which states that monoamines are somehow depleted (Figure 6-34B), and when boosted with effective antidepressants relieve depression (Figure 7-8). One problem for the monoamine hypothesis, however, is that the action of antidepressants at monoamine transporters can raise monoamine levels quite rapidly in some brain areas, and certainly sooner than the antidepressant clinical effects occur in patients weeks later (Figure 7-8). How could immediate changes in neurotransmitter levels caused by antidepressants be linked to clinical actions that are much later in time? The answer may be that the acute increases in neurotransmitter levels cause adaptive changes in neurotransmitter receptor sensitivity in a delayed time course consistent with the onset of clinical antidepressant actions (Figure 7-8). Specifically, acutely enhanced synaptic levels of neurotransmitter (Figure 7-9A) could lead to adaptive downregulation and desensitization of postsynaptic neurotransmitter receptors over time (Figure 7-9B).
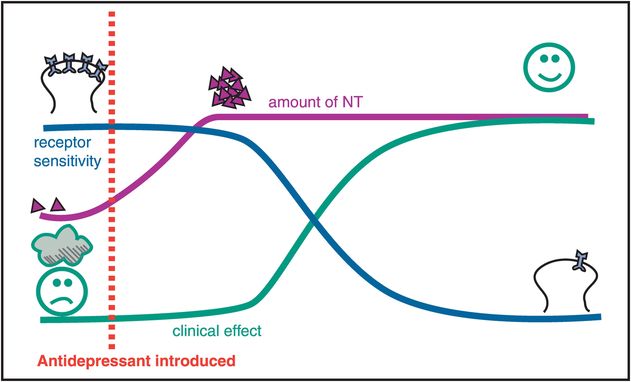
Figure 7-8. Time course of antidepressant effects. This figure depicts the different time courses for three effects of antidepressant drugs – namely, clinical changes, neurotransmitter (NT) changes, and receptor sensitivity changes. Specifically, the amount of NT changes relatively rapidly after an antidepressant is introduced. However, the clinical effect is delayed, as is the desensitization, or downregulation, of neurotransmitter receptors. This temporal correlation of clinical effects with changes in receptor sensitivity has given rise to the hypothesis that changes in neurotransmitter receptor sensitivity may actually mediate the clinical effects of antidepressant drugs. These clinical effects include not only antidepressant and anxiolytic actions but also the development of tolerance to the acute side effects of antidepressant drugs.
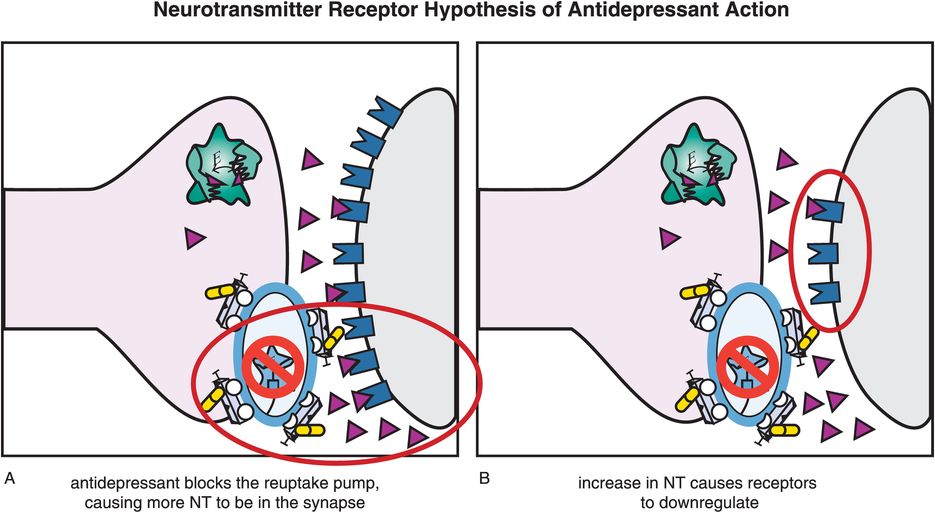
Figure 7-9. Neurotransmitter receptor hypothesis of antidepressant action. Although antidepressants cause an immediate increase in monoamines, they do not have immediate therapeutic effects. This may be explained by the monoamine receptor hypothesis of depression, which states that depression is caused by upregulation of monoamine receptors; thus antidepressant efficacy would be related to downregulation of those receptors, as shown here. (A) When an antidepressant blocks a monoamine reuptake pump, this causes more neurotransmitter (NT) (in this case, norepinephrine) to accumulate in the synapse. (B) The increased availability of NT ultimately causes receptors to downregulate. The time course of receptor adaptation is consistent both with the delayed clinical effects of antidepressants and with development of tolerance to antidepressant side effects.
This concept of antidepressants causing changes in neurotransmitter receptor sensitivity is also consistent with the neurotransmitter receptor hypothesis of depression causing upregulation of neurotransmitter receptors in the first place (Figure 7-9A). Thus, antidepressants theoretically reverse this pathological upregulation of receptors over time (Figure 7-9B). Furthermore, the time course of receptor adaptation fits both with the onset of therapeutic effects and with the onset of tolerance to many side effects. Different receptors likely mediate these different actions, but both the onset of therapeutic action and the onset of tolerance to side effects may occur with the same delayed time course.
Adaptive changes in receptor number or sensitivity are likely the result of alterations in gene expression (Figure 7-10). This may include not only turning off the synthesis of neurotransmitter receptors, but also increasing the synthesis of various neurotrophic factors such as BDNF (brain-derived neurotrophic factor) (Figure 7-10), as also discussed in Chapter 6 and illustrated in Figures 6-36 through 6-38. Such mechanisms may apply broadly to all effective antidepressants, and may provide a final common pathway for the action of antidepressants.
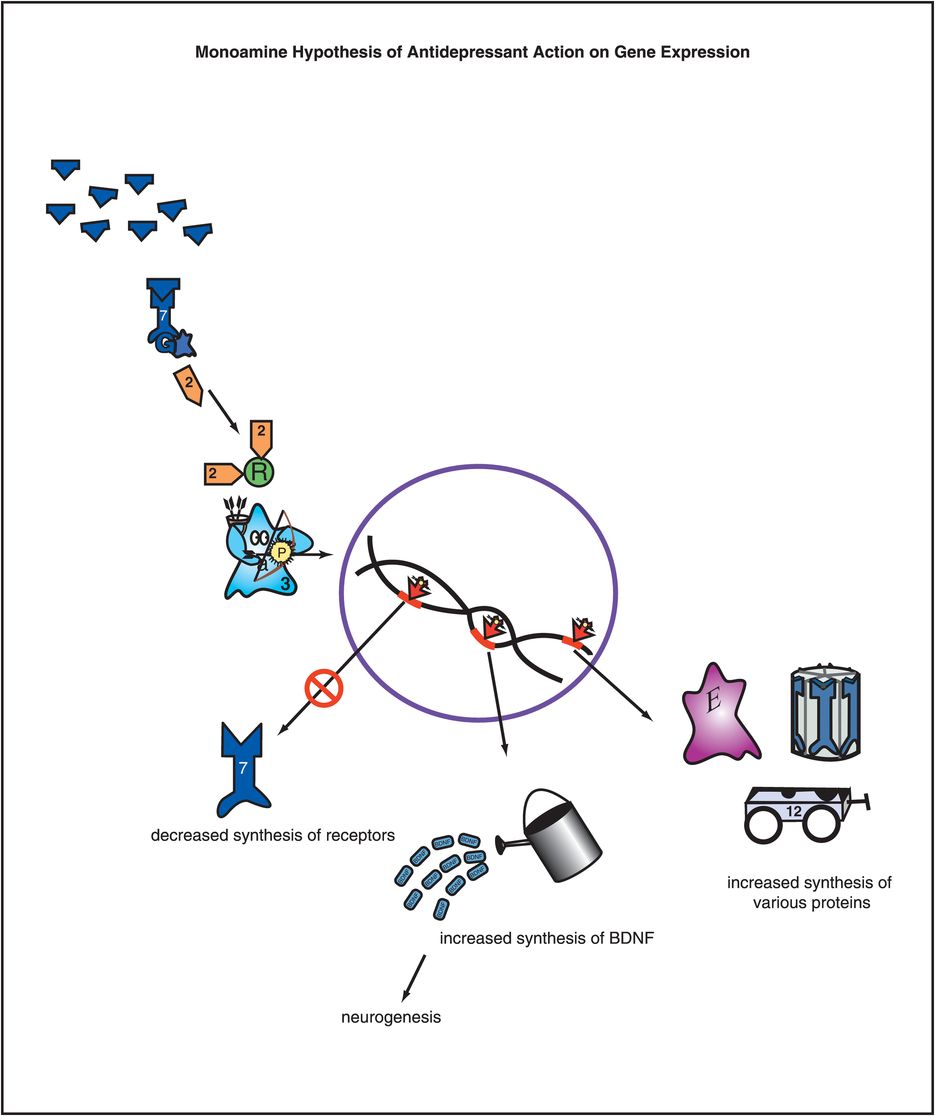
Figure 7-10. Monoamine hypothesis of antidepressant action on gene expression. Adaptations in receptor number or sensitivity are likely due to alterations in gene expression, as shown here. The neurotransmitter at the top is presumably increased by an antidepressant. The cascading consequence of this is ultimately to change the expression of critical genes in order to effect an antidepressant response. This includes downregulating some genes so that there is decreased synthesis of receptors as well as upregulating other genes so that there is increased synthesis of critical proteins, such as brain-derived neurotrophic factor (BDNF).
Selective serotonin reuptake inhibitors (SSRIs)
Rarely has a class of drugs transformed a field as dramatically as have the SSRIs transformed clinical psychopharmacology. Some estimate that SSRI prescriptions in the US alone occur at the rate of six prescriptions per second, 24/7, year round. Already prominent in Europe, SSRIs are now entering Japan and all across Asia, with increasing use throughout the entire world. Clinical indications for the use of SSRIs range far beyond major depressive disorder, especially to a number of anxiety disorders, and also to premenstrual dysphoric disorder, eating disorders, and beyond. There are six principal agents in this group that all share the common property of serotonin reuptake inhibition, and thus they all belong to the same drug class, known as SSRIs. However, each of these six drugs also has unique pharmacological properties that allow them to be distinguished from each other. First, we will discuss what these six drugs share in common, and then we will explore their distinctive individual properties that allow sophisticated prescribers to match specific drug profiles to individual patient symptom profiles.
What the six SSRIs have in common
All six SSRIs have the same major pharmacologic feature in common: selective and potent inhibition of serotonin reuptake, also known as inhibition of the serotonin transporter or SERT (Figure 7-11). This simple concept was introduced in Chapter 5 and illustrated in Figure 5-14 and is shown here in Figure 7-12. Although the action of SSRIs at the presynaptic axon terminal has classically been emphasized (Figure 7-12), it now appears that events occurring at the somatodendritic end of the serotonin neuron (near the cell body) may be more important in explaining the therapeutic actions of the SSRIs (Figures 7-13 through 7-17). That is, in the depressed state, the monoamine hypothesis of depression states that serotonin may be deficient, both at presynaptic somatodendritic areas near the cell body (on the left in Figure 7-13) and in the synapse itself near the axon terminal (on the right in Figure 7-13). The neurotransmitter receptor hypothesis proposes that monoamine receptors may be upregulated as shown in Figure 7-13, representing the depressed state before treatment. Neuronal firing rates may also be dysregulated in depression, contributing to regional abnormalities in information processing, and the development of specific symptoms depending upon the region affected, as discussed in Chapter 6 and shown in Figures 6-33 and 6-45.
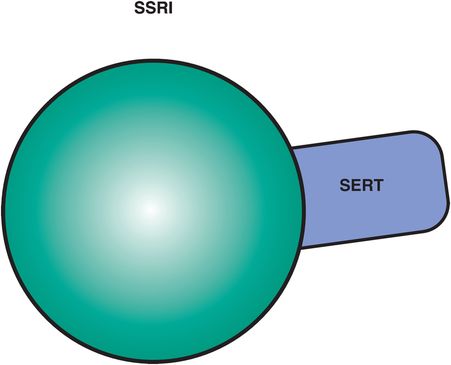
Figure 7-11. Selective serotonin reuptake inhibitors. Shown here is an icon depicting the core feature of selective serotonin reuptake inhibitors (SSRIs), namely serotonin reuptake inhibition. Although the agents in this class have unique pharmacological profiles, they all share the common property of serotonin transporter (SERT) inhibition.
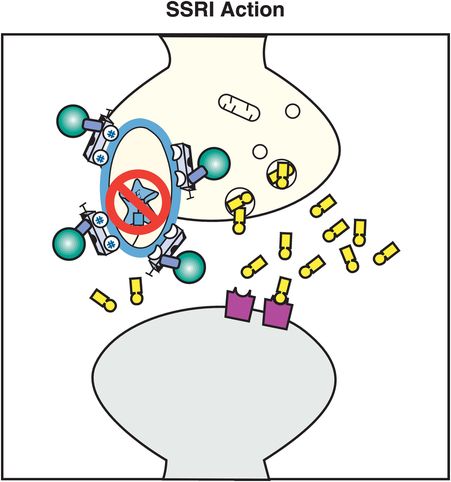
Figure 7-12. SSRI action. In this figure, the serotonin reuptake inhibitor (SRI) portion of the SSRI molecule is shown inserted into the serotonin reuptake pump (the serotonin transporter, or SERT), blocking it and causing an antidepressant effect.
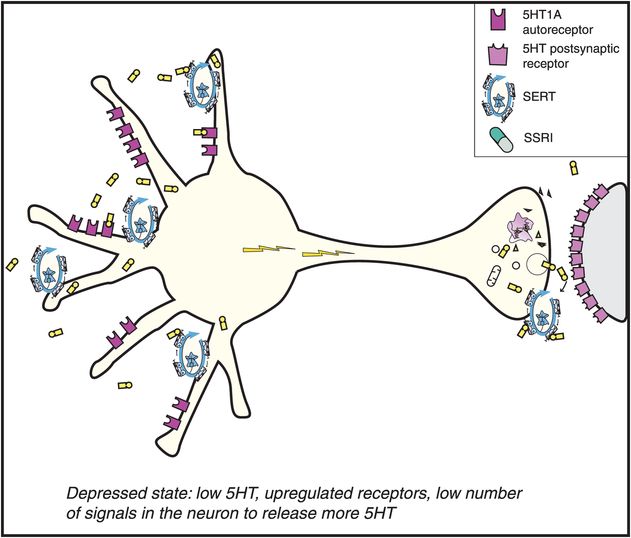
Figure 7-13. Mechanism of action of selective serotonin reuptake inhibitors (SSRIs), part 1. Depicted here is a serotonin (5HT) neuron in a depressed patient. In depression, the 5HT neuron is conceptualized as having a relative deficiency of the neurotransmitter 5HT. Also, the number of 5HT receptors is upregulated, including presynaptic 5HT1A autoreceptors as well as postsynaptic 5HT receptors.
When an SSRI is given acutely, it is well known that 5HT rises due to blockade of SERT. What is somewhat surprising, however, is that blocking the presynaptic SERT does not immediately lead to a great deal of serotonin in many synapses. In fact, when SSRI treatment is initiated, 5HT rises to much greater levels at the somatodendritic area located in the midbrain raphe (on the left in Figure 7-14) due to blockade of SERTs there, rather than in the areas of the brain where the axons terminate (on the right in Figure 7-14).
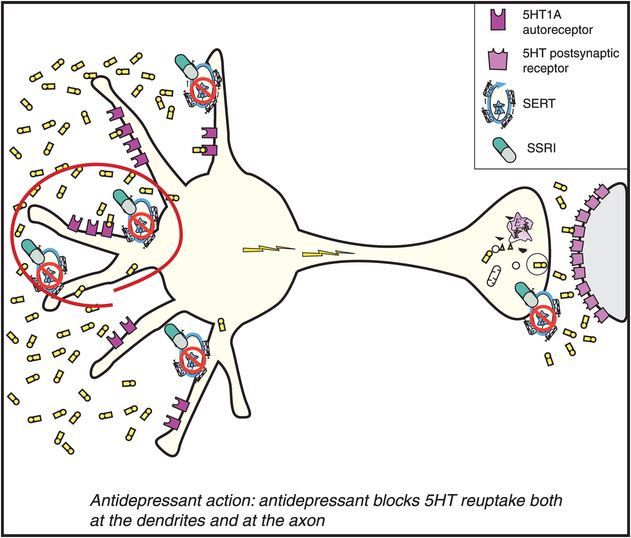
Figure 7-14. Mechanism of action of selective serotonin reuptake inhibitors (SSRIs), part 2. When an SSRI is administered, it immediately blocks the serotonin reuptake pump (see icon of an SSRI drug capsule blocking the reuptake pump, or serotonin transporter [SERT]). However, this causes serotonin to increase initially only in the somatodendritic area of the serotonin neuron (left) and not very much in the axon terminals (right).
The somatodendritic area of the serotonin neuron is therefore where 5HT increases first (on the left in Figure 7-14). Serotonin receptors in this brain area have 5HT1A pharmacology as discussed in Chapter 5 and illustrated in Figure 5-25. When serotonin levels rise in the somatodendritic area, they stimulate nearby 5HT1A autoreceptors (also on the left in Figure 7-14). These immediate pharmacologic actions obviously cannot explain the delayed therapeutic actions of the SSRIs. However, these immediate actions may explain the side effects that are caused by the SSRIs when treatment is initiated.
Over time, the increased 5HT acting at the somatodendritic 5HT1A autoreceptors causes them to downregulate and become desensitized (on the left in Figure 7-15). This desensitization occurs because the increase in serotonin is recognized by these presynaptic 5HT1A receptors, and this information is sent to the cell nucleus of the serotonin neuron. The genome’s reaction to this information is to issue instructions that cause these same receptors to become desensitized over time. The time course of this desensitization correlates with the onset of therapeutic actions of the SSRIs.
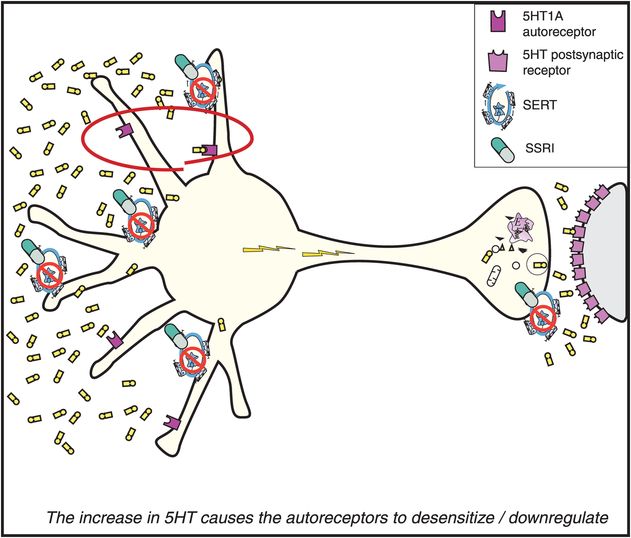
Figure 7-15. Mechanism of action of selective serotonin reuptake inhibitors (SSRIs), part 3. The consequence of serotonin increasing in the somatodendritic area of the serotonin (5HT) neuron, as depicted in Figure 7-14, is that the somatodendritic 5HT1A autoreceptors desensitize or downregulate (red circle).
Once the 5HT1A somatodendritic autoreceptors are desensitized, 5HT can no longer effectively turn off its own release. Since 5HT is no longer inhibiting its own release, the serotonin neuron is therefore disinhibited (Figure 7-16). This results in a flurry of 5HT release from axons and an increase in neuronal impulse flow (shown as lightning in Figure 7-16 and release of serotonin from the axon terminal on the right). This is just another way of saying the serotonin release is “turned on” at the axon terminals. The serotonin that now pours out of the various projections of serotonin pathways in the brain is what theoretically mediates the various therapeutic actions of the SSRIs.
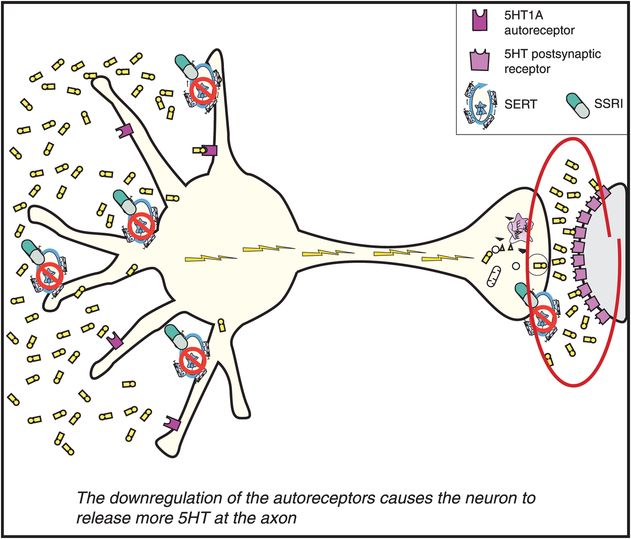
Figure 7-16. Mechanism of action of selective serotonin reuptake inhibitors (SSRIs), part 4. Once the somatodendritic receptors downregulate, as depicted in Figure 7-15, there is no longer inhibition of impulse flow in the serotonin (5HT) neuron. Thus, neuronal impulse flow is turned on. The consequence of this is release of 5HT in the axon terminal (red circle). However, this increase is delayed as compared with the increase of 5HT in the somatodendritic areas of the 5HT neuron, depicted in Figure 7-14. This delay is the result of the time it takes for somatodendritic 5HT to downregulate the 5HT1A autoreceptors and turn on neuronal impulse flow in the 5HT neuron. This delay may explain why antidepressants do not relieve depression immediately. It is also the reason why the mechanism of action of antidepressants may be linked to increasing neuronal impulse flow in 5HT neurons, with 5HT levels increasing at axon terminals before an SSRI can exert its antidepressant effects.
While the presynaptic somatodendritic 5HT1A autoreceptors are desensitizing (Figure 7-15), serotonin is building up in synapses (Figure 7-16), and causes the postsynaptic serotonin receptors to desensitize as well (on the right in Figure 7-17). These various postsynaptic serotonin receptors in turn send information to the cell nucleus of the postsynaptic neuron that serotonin is targeting (on the far right of Figure 7-17). The reaction of the genome in the postsynaptic neuron is also to issue instructions to downregulate or desensitize these receptors as well. The time course of this desensitization correlates with the onset of tolerance to the side effects of the SSRIs (Figure 7-17).
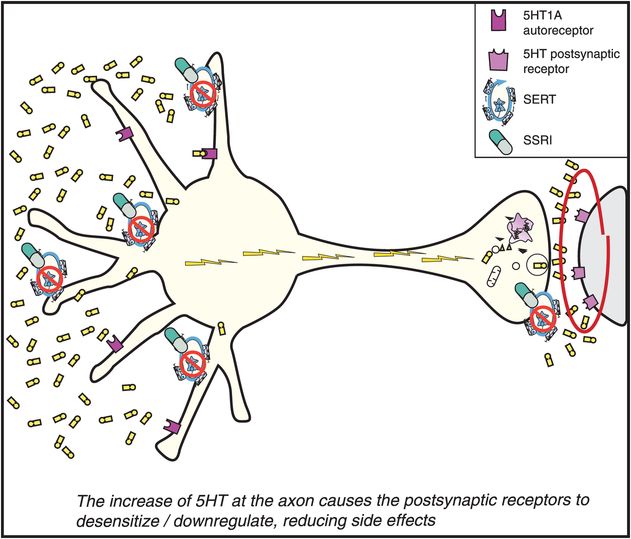
Figure 7-17. Mechanism of action of selective serotonin reuptake inhibitors (SSRIs), part 5. Finally, once the SSRIs have blocked the reuptake pump (or serotonin transporter [SERT] in Figure 7-14), increased somatodendritic serotonin (5HT) (Figure 7-14), desensitized somatodendritic 5HT1A autoreceptors (Figure 7-15), turned on neuronal impulse flow (Figure 7-16), and increased release of 5HT from axon terminals (Figure 7-16), the final step (shown here) may be the desensitization of postsynaptic 5HT receptors. This desensitization may mediate the reduction of side effects of SSRIs as tolerance develops.
This theory thus suggests a pharmacological cascading mechanism whereby the SSRIs exert their therapeutic actions: namely, powerful but delayed disinhibition of serotonin release in key pathways throughout the brain. Furthermore, side effects are hypothetically caused by the acute actions of serotonin at undesirable receptors in undesirable pathways. Finally, side effects may attenuate over time by desensitization of the very receptors that mediate them.
Unique properties of each SSRI: the not-so-selective serotonin reuptake inhibitors
Although the six SSRIs clearly share the same mechanism of action, individual patients often react very differently to one SSRI versus another. This is not generally observed in large clinical trials, where mean group differences between two SSRIs either in efficacy or in side effects are very difficult to document. Rather, such differences are seen by prescribers treating patients one at a time, with some patients experiencing a therapeutic response to one SSRI and not another, and other patients tolerating one SSRI and not another.
If blockade of SERT explains the shared clinical and pharmacological actions of SSRIs, what explains their differences? Although there is no generally accepted explanation that accounts for the commonly observed clinical phenomena of different efficacy and tolerability of various SSRIs in individual patients, it makes sense to consider those pharmacologic characteristics of the six SSRIs that are not shared with each other as candidates to explain the broad range of individual patient reactions to different SSRIs (Figures 7-18 through 7-23). Each SSRI has secondary pharmacologic actions other than SERT blockade, and no two SSRIs have identical secondary pharmacological characteristics. Whether these secondary binding profiles can account for the differences in efficacy and tolerability in individual patients remains to be proven. However, it does lead to provocative hypothesis generation and gives a rational basis for psychopharmacologists trying more than one of these agents rather than thinking “they are all the same.” Sometimes only an empiric trial of different SSRIs will lead to the best match of drug to an individual patient.
Fluoxetine: an SSRI with 5HT2C antagonist properties
This SSRI also has 5HT2C antagonist actions that may explain many of its unique clinical properties (Figure 7-18). 5HT2C antagonism is explained in Chapter 5 and illustrated in Figures 5-52A and 5-52B. Other antidepressants with 5HT2C antagonist properties include mirtazapine and agomelatine; several atypical antipsychotics including quetiapine with proven antidepressant properties, as well as olanzapine, asenapine, and clozapine, also have potent 5HT2C antagonist actions. Blocking serotonin action at 5HT2C receptors disinhibits (i.e., enhances) release of both NE and DA (Figure 5-52B). 5HT2C antagonism may contribute not only to fluoxetine’s therapeutic actions but also to its tolerability profile.
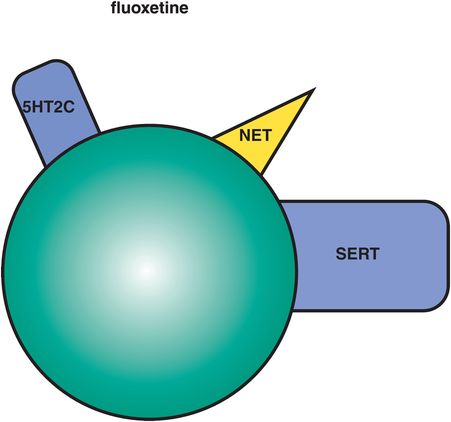
Figure 7-18. Fluoxetine. In addition to serotonin reuptake inhibition, fluoxetine has norepinephrine reuptake inhibition (NRI) and serotonin 2C (5HT2C) antagonist actions. Fluoxetine’s activating effects may be due to its actions at 5HT2C receptors. Norepinephrine reuptake inhibition may be clinically relevant only at very high doses. Fluoxetine is also an inhibitor at CYP 2D6 and 3A4.
The good news about 5HT2C antagonism may be that it is generally activating, and many patients, even from the first dose, detect an energizing and fatigue-reducing effect of fluoxetine, with improvement in concentration and attention as well. This mechanism is perhaps best matched to depressed patients with reduced positive affect, hypersomnia, psychomotor retardation, apathy, and fatigue (Figure 6-46). Fluoxetine is also approved in some countries in combination with olanzapine for treatment-resistant unipolar depression and for bipolar depression. Since olanzapine also has 5HT2C antagonist actions (Figure 5-46), it may be that adding together the 5HT2C antagonist actions of both drugs could theoretically lead to further enhanced DA and NE release in cortex to mediate the antidepressant actions of this combination. 5HT2C antagonism may also contribute to the anti-bulimia effect of higher doses of fluoxetine, the only SSRI approved for the treatment of this eating disorder.
The bad news about 5HT2C antagonism is that it can be activating, so the 5HT2C antagonist actions of fluoxetine may contribute to this agent being sometimes less well matched to patients with agitation, insomnia, and anxiety, who may experience unwanted activation and even a panic attack if given an agent that further activates them.
Other unique properties of fluoxetine (Figure 7-18) are weak NE reuptake blocking properties that may become clinically relevant at very high doses. Fluoxetine has a long half-life (2–3 days), and its active metabolite an even longer half-life (2 weeks). The long half-life is advantageous in that it seems to reduce the withdrawal reactions that are characteristic of sudden discontinuation of some SSRIs, but it also means that it takes a long time to clear the drug and its active metabolite after discontinuing fluoxetine, and prior to starting another agent such as a monoamine oxidase inhibitor (MAOI). Fluoxetine is available not only as a once-daily formulation, but also as a once-weekly oral dosage formulation.
Sertraline: an SSRI with dopamine transporter (DAT) inhibition and σ1 binding
This SSRI has two candidate mechanisms that distinguish it: dopamine transporter (DAT) inhibition and sigma-1 (σ1) receptor binding (Figure 7-19). The DAT inhibitory actions are controversial since they are weaker than the SERT inhibitory actions, thus leading some experts to suggest that there is not sufficient DAT occupancy by sertraline to be clinically relevant. However, as will be discussed later in the section on norepinephrine–dopamine reuptake inhibitors (NDRIs), it is not clear that high degrees of DAT occupancy are necessary or even desirable in order to contribute to antidepressant actions. That is, perhaps only a small amount of DAT inhibition is sufficient to cause improvement in energy, motivation, and concentration, especially when added to another action such as SERT inhibition. In fact, high-impact DAT inhibition is the property of reinforcing stimulants, including cocaine and methylphenidate, and would not generally be desired in an antidepressant.
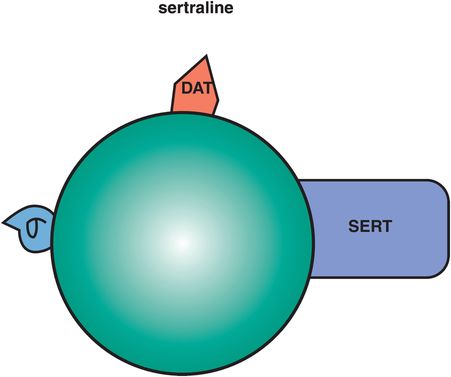
Figure 7-19. Sertraline. Sertraline has dopamine reuptake inhibition (DRI) and σ1 receptor binding in addition to serotonin reuptake inhibition (SRI). The clinical relevance of sertraline’s DRI is unknown, although it may improve energy, motivation, and concentration. Its σ properties may contribute to anxiolytic actions and may also be helpful in patients with psychotic depression.
Anecdotally, clinicians have observed the mild and desirable activating actions of sertraline in some patients with “atypical depression,” improving symptoms of hypersomnia, low energy, and mood reactivity. A favorite combination of some clinicians for depressed patients is to add bupropion to sertraline (i.e., Wellbutrin to Zoloft, sometimes called “Well-oft”), adding together the weak DAT inhibitory properties of each agent. Clinicians have also observed the over-activation of some patients with panic disorder by sertraline, thus requiring slower dose titration in some patients with anxiety symptoms. All of these actions of sertraline are consistent with weak DAT inhibitory actions of sertraline contributing to its clinical portfolio of actions.
The σ1 actions of sertraline are not well understood, but might contribute to its anxiolytic effects and especially to its effects in psychotic and delusional depression, where sertraline may have advantageous therapeutic effects compared to some other SSRIs. These σ1 actions could theoretically contribute both to anxiolytic actions and to antipsychotic actions, as will be discussed further in the section on fluvoxamine below.
Paroxetine: an SSRI with muscarinic anticholinergic and norepinephrine transporter (NET) inhibitory actions
This SSRI is preferred by many clinicians for patients with anxiety symptoms. It tends to be more calming, even sedating, early in treatment compared to the more activating actions of both fluoxetine and sertraline discussed above. Perhaps the mild anticholinergic actions of paroxetine contribute to this clinical profile (Figure 7-20). Paroxetine also has weak NET (norepinephrine transporter) inhibitory properties, which could contribute to its efficacy in depression, especially at high doses. The advantages of dual serotonin plus norepinephrine reuptake inhibiting properties, or SNRI actions, are discussed below in the section on SNRIs. It is possible that weak to moderate NET inhibition of paroxetine may contribute importantly to its antidepressant actions.
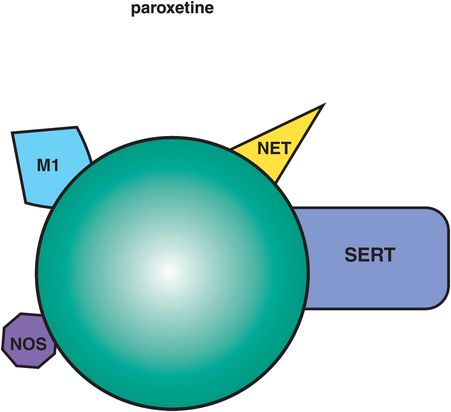
Figure 7-20. Paroxetine. In addition to serotonin reuptake inhibition (SRI), paroxetine has mild anticholinergic actions (M1), which can be calming or possibly sedating, weak norepinephrine reuptake inhibition (NRI), which may contribute to further antidepressant actions, and inhibition of the enzyme nitric oxide synthetase (NOS), which may contribute to sexual dysfunction. Paroxetine is also a potent inhibitor of CYP 2D6.
Paroxetine inhibits the enzyme nitric oxide synthetase, which could theoretically contribute to sexual dysfunction especially in men. Paroxetine is also notorious for causing withdrawal reactions upon sudden discontinuation with symptoms such as akathisia, restlessness, gastrointestinal symptoms, dizziness, and tingling, especially when suddenly discontinued from long-term high-dose treatment. This is possibly due not only to SERT inhibition properties, since all SSRIs can cause discontinuation reactions, but also to additional contributions from anticholinergic rebound when paroxetine is rapidly discontinued. Paroxetine is available in a controlled-release formulation, which may mitigate some of its side effects, including discontinuation reactions.
Fluvoxamine: an SSRI with σ1 receptor binding properties
This SSRI was among the first to be launched for the treatment of depression worldwide, but was never officially approved for depression in the US, so has been considered more of an agent for the treatment of obsessive–compulsive disorder and anxiety in the US. A unique binding property of fluvoxamine, like sertraline, is its interaction at σ1 sites, but this action is more potent for fluvoxamine than for sertraline (Figure 7-21). The physiological function of σ1 sites is still a mystery, and thus sometimes called the “sigma enigma,” but has been linked to both anxiety and psychosis. Although it is not entirely clear how to define an agonist or antagonist at σ1 sites, recent studies suggest that fluvoxamine may be an agonist at σ1 receptors, and that this property may contribute an additional pharmacologic action to help explain fluvoxamine’s well-known anxiolytic properties. Fluvoxamine also has shown therapeutic activity in both psychotic and delusional depression, where it, like sertraline, may have advantages over other SSRIs.
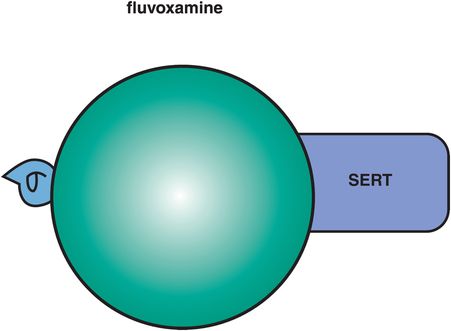
Figure 7-21. Fluvoxamine. Fluvoxamine’s secondary properties include actions at σ1 receptors, which may be anxiolytic as well as beneficial for psychotic depression, and inhibition of CYP 1A2 and 3A4.
Fluvoxamine is now available as a controlled-release formulation which makes once-a-day administration possible, unlike immediate-release fluvoxamine, whose shorter half-life often requires twice-daily administration. In addition, recent trials of controlled-release fluvoxamine show impressive remission rates in both obsessive–compulsive disorder and social anxiety disorder, as well as possibly less peak dose sedation.
Citalopram: an SSRI with a “good” and a “bad” enantiomer
This SSRI is comprised of two enantiomers, R and S, one of which is the mirror image of the other (Figure 7-22). The mixture of these enantiomers is known as racemic citalopram, or commonly just as citalopram, and it has mild antihistaminic properties that reside in the R enantiomer (Figure 7-22). Racemic citalopram is generally one of the better-tolerated SSRIs, and has favorable findings in the treatment of depression in the elderly, but has a somewhat inconsistent therapeutic action at the lowest dose, often requiring dose increase to optimize treatment. However, dose increase is limited due to the potential for QTc prolongation. These findings all suggest that it is not favorable for citalopram to contain the R enantiomer. In fact, some pharmacologic evidence suggests that the R enantiomer may be pharmacologically active at SERT in a manner that does not inhibit SERT but actually interferes with the ability of the active S enantiomer to inhibit SERT. This could lead to reduced inhibition of SERT, reduced synaptic 5HT, and possibly reduced net therapeutic actions, especially at low doses.
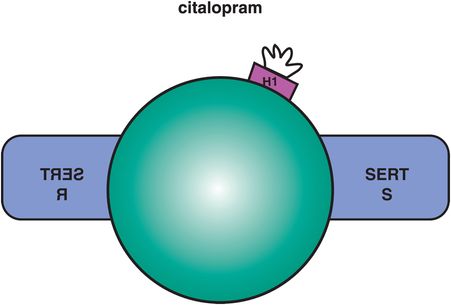
Figure 7-22. Citalopram. Citalopram consists of two enantiomers, R and S. The R enantiomer has weak antihistamine properties and is a weak inhibitor of CYP 2D6.
Escitalopram: the quintessential SSRI
The solution to improving the properties of racemic citalopram is to remove the unwanted R enantiomer. The resulting drug is known as escitalopram, as it is composed of only the pure active S enantiomer (Figure 7-23). This maneuver appears to remove the antihistaminic properties, and there are no higher dose restrictions to avoid QTc prolongation. In addition, removal of the potentially interfering R isomer makes the lowest dose of escitalopram more predictably efficacious. Escitalopram is therefore the SSRI for which pure SERT inhibition is most likely to explain almost all of its pharmacologic actions. Escitalopram is considered perhaps the best-tolerated SSRI, with the fewest CYP-mediated drug interactions.
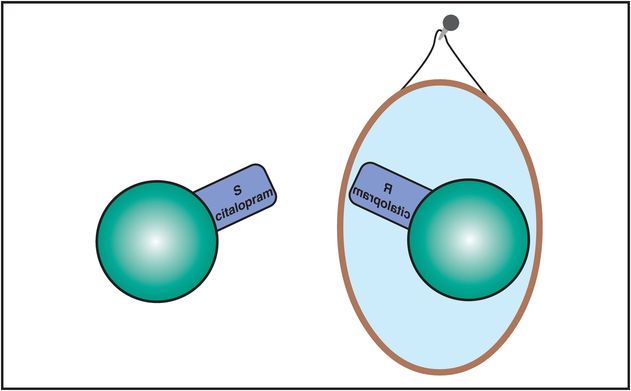
Figure 7-23. Escitalopram. The R and S enantiomers of citalopram are mirror images of each other but have slightly different clinical properties. The R enantiomer is the one with weak antihistamine properties and weak inhibition of CYP 2D6, while the S enantiomer does not have these properties. The R and S enantiomers may also differ in their effects at the serotonin transporter. The S enantiomer of citalopram has been developed and marketed as the antidepressant escitalopram.
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


