a. 100%
b. 75%
c. 50%
d. 25%
e. Virtually 0
349. A 3-year-old Caucasian boy is evaluated by an ophthalmologist and is found to have retinitis pigmentosa, a disorder characterized by pigmentary granules in the retina and progressive vision loss. The pedigree below is obtained and the family comes in for counseling. What is the risk for individual II-2 of having an affected child if he impregnates an unrelated female?
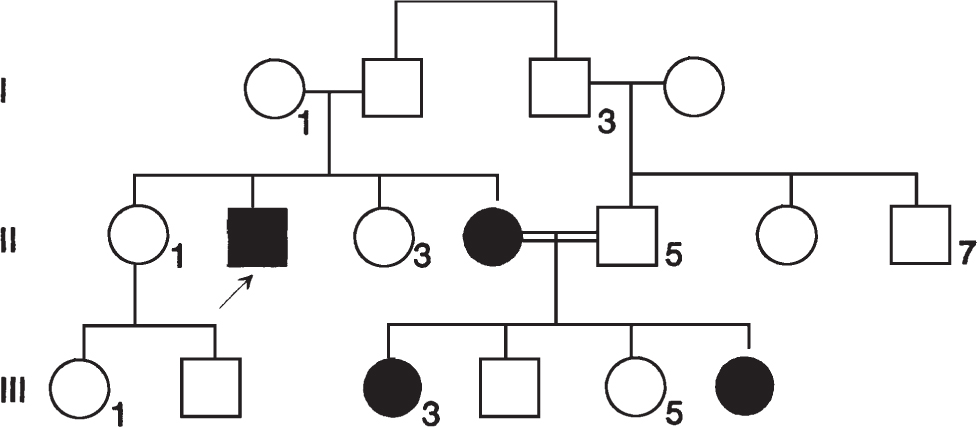
a. 100%
b. 75%
c. 50%
d. 25%
e. Virtually 0
350. A 6-month-old Caucasian boy of Ashkenazi Jewish origin plateaus in his development, exhibits a “startle” response to hand-claps, and is noted to have a central red area in his retina surrounded by white (cherry red spot). His physician suspects autosomal recessive Tay-Sachs disease (MIM*272800) and initiates evaluation that demonstrates deficiency of the lysosomal enzyme hexaminidase A. This deficiency leads to accumulation of complex glycolipids in brain and produces a ring of white, lipid-infiltrated cells around normal central retina (the macula). What is the risk that the grandmother of an affected child is a carrier (heterozygote) for Tay-Sachs disease?
a. 100%
b. 67%
c. 50%
d. 25%
e. Virtually 0
351. A 1-month-old Caucasian male infant is being considered for adoption, and his older half sister is known to have developed hydrocephalus, an accumulation of cerebrospinal fluid in the brain ventricles. Hydrocephalus is a multifactorial disorder, and the prospective parents wish to know the chance the boy will develop hydrocephalus. In order to estimate this risk, the physician must determine what proportion of genes the brother and the half sister have in common. What is this proportion?
a. 1
b. ½
c. ¼
d. 1/8
e. 1/16
352. A 66-year-old African American female is diagnosed with Parkinson disease (PD-MIM*168601) and there are no other cases in her family. She requests genetic counseling and/or testing to assess risks that her middleaged children will develop parkinsonism. Her physician explains that 1% of people over 50 may contract the disease, that rare families exhibit autosomal dominant inheritance, and that usual risks are increased three-to fourfold for first-degree relatives of affected individuals. Based on this information, which of the following options provide appropriate genetic counseling for this patient?
a. Likely multifactorial determination in your family with a 3% to 4% risk for your middle-aged children to develop PD
b. Likely multifactorial determination in your family with a 1% risk for your middle-aged children to develop PD
c. Likely autosomal dominant inheritance in your family with a 50% risk for your middle-aged children to develop PD
d. Likely autosomal dominant inheritance in your family with a 25% risk for your middle-aged children to develop PD
e. Likely multifactorial determination in your family with a 10% risk for your middle-aged children to develop PD
353. In the operating room, a 5-year-old Hispanic boy receives succinylcholine as a muscle relaxant to facilitate intubation and anesthesia. The operation proceeds until it is time for recovery, when the child does not begin breathing. A hurried discussion with the father discloses no additional problems in the family, but he does say that he and his wife are first cousins. Which of the following is the most likely possibility?
a. An autosomal dominant disorder that interferes with succinylcholine metabolism
b. An autosomal recessive disorder that interferes with succinylcholine metabolism
c. An X-linked disorder that interferes with succinylcholine metabolism
d. A lethal gene transmitted through consanguinity that affects the respiratory system
e. Mismanagement of halothane anesthesia during the operation
354. Pharmacogenetics, or the study of drug-induced disease due to genetic variation, is receiving increased attention particularly with regard to population or at least preoperative screening. The frequency of heterozygotes for variant butyrylcholinesterase (BChE) alleles in Caucasians is about 4 per 100, implying an incidence of individuals with potential for severe apnea of which of the following?
a. 1 in 5000
b. 1 in 2500
c. 1 in 1250
d. 1 in 500
e. 1 in 50
355. A 6-month-old Caucasian girl of Ashkenazi Jewish background seems to plateau in development after a normal gestation, delivery, and early infancy. She rolled over well with smiling and good interaction but became less active and does not maintain a sit. Her pediatrician notes low muscle tone and claps her hands to elicit an exaggerated extension of her arms (Moro reflex—enhanced startle response). Ophthalmologic examination reveals a central red area of the retina surrounded by white tissue (cherry red spot). A diagnosis of lipid storage disease (neurolipidosis) is suspected. If the diagnosis is correct, what is the risk that the next child of these parents will be affected with the same disease?
a. ½
b. ¼
c. ¾
d. 1/12
e. 1/24
356. The cause of Tay-Sachs disease (MIM*272800) is best described by which of the following?
a. Excess of a lysosomal enzyme in blood due to defective uptake
b. Deficiency of a lysosomal enzyme that digests proteoglycans
c. Deficiency of a membrane receptor that takes up proteoglycans
d. Deficiency of a mitochondrial enzyme that degrades glycogen
e. Deficiency of a mitochondrial triglyceride lipase
357. The frequency of carriers of Tay-Sachs disease (MIM*272800) in Ashkenazi Jewish populations is 1 in 30. Some Jewish communities offer carrier testing for young adults so they will know their status before marriage. A known carrier female becomes involved with an exchange student from Russia who also is Ashkenazi Jewish but who has not had carrier testing. What is the chance that a child of this union will have Tay-Sachs disease?
a. 1 in 2 1/120
b. 1 in 15 1/240
c. 1 in 30 1/3600
d. 1 in 60 1/9000
e. 1 in 120 1/36,000
358. A couple decide to have prenatal diagnosis because their previous child has Tay-Sachs disease (MIM*272800). Which of the following prenatal diagnostic techniques is optimal for fetal diagnosis?
a. Chorionic villus sampling (CVS)
b. Percutaneous umbilical blood sampling
c. Amniotic fluid α-fetoprotein levels
d. Maternal serum α-fetoprotein (MSAFP)
e. Fetal x-rays
359. A 2-year-old child is hospitalized for evaluation of poor growth and low muscle tone. The most striking physical finding is unruly, “kinky” hair, but the child also has increased joint laxity and thin skin. Which of the following laboratory findings is most likely?
a. High ceruloplasmin
b. High tissue copper
c. Low serum iron
d. Low saturation of transferrin
e. Low serum haptoglobin
360. A male child presents with delayed development and scarring of his lips and hands. His parents have restrained him because he obsessively chews on his lips and fingers. Which of the following is likely to occur in this child?
a. Increased levels of 5-phosphoribosyl-1-pyrophosphate (PRPP)
b. Decreased purine synthesis
c. Decreased levels of uric acid
d. Increased levels of hypoxanthine-guanosine phosphoribosyl transferase (HGPRT)
e. Glycogen storage
361. The figure below shows a pedigree that includes individuals with Charcot-Marie-Tooth disease (CMT), a neurologic disorder that produces dysfunction of the distal extremities with characteristic footdrop. If individual III-4 becomes pregnant, what is her risk of having a child with CMT?
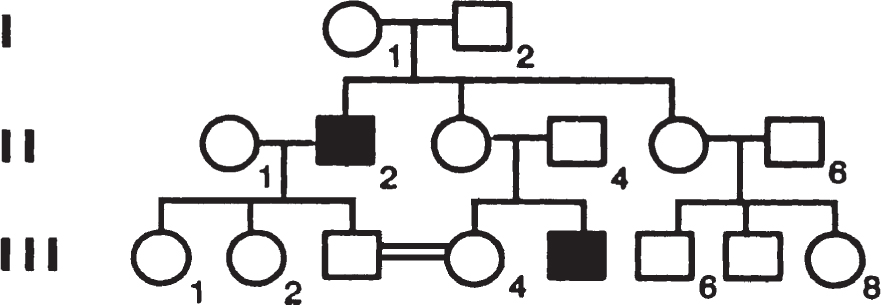
a. ½
b. ¼
c. 1/8
d. 1/16
e. Virtually 0
362. A child with severe epilepsy, autistic behavior, and developmental delay has characteristics of a condition known as Angelman syndrome (MIM*105830). Because of the syndromic nature of the disorder and the developmental delay, a karyotype is performed that shows a missing band on one chromosome 15. Which of the following best describes this abnormality?
a. Interstitial deletion of 15
b. Terminal deletion of 15
c. Pericentric inversion of 15
d. Paracentric inversion of 15
e. 15q–
Neurosensory and Neuromuscular Systems—Neurology, Ophthalmology, Otolaryngology
Answers
327. The answer is e. (Murray, pp 354-362. Scriver, pp 3-45. Lewis, pp 127-128.) DNA methylation occurs mainly at CpG dinucleotides that often cluster in the upstream promoter regions of genes (CpG islands). Incorrect answers reflect double crossovers at meiosis that substitutes a normal allele for a mutant allele (gene conversion—answer a), reverse transcriptases copying intronless mRNA into complementary DNAs (cDNAs) that integrate into the genome as pseudogenes (answer c), gene rearrangement to unite variable, joining, and constant regions of immunoglobulin genes for expression of a unique antibody (answer d), or unequal crossing-over between sister chromatids is thought to be an important mechanism for variation in copy number within gene clusters (answer b). Chromosomes subject to genomic imprinting may have different DNA methylation patterns on the homologue transmitted from mother versus that transmitted by father, producing balanced expression that is disrupted in disorders such as Prader-Willi syndrome (MIM*176270).
328. The answer is c. (Murray, pp 361-362. Scriver, pp 2415-2512. Lewis, pp 98-100.) Mitochondrial DNA is similar to bacterial DNA in structure as a circular, double-stranded molecule of 16,569 bp (eliminates answers a and d positing single-stranded DNA, b and e positing linear DNA). Present in 500 to 1000 copies per cell, it comprises 1% of cellular DNA and encodes 13 peptides of the respiratory chain compared to 54 encoded by nuclear DNA. Mitochondrial DNA encodes unique ribosomal and transfer RNAs, has some unique coding properties (UGA stop codon read as tryptophan), and exhibits a 5- to 10-fold higher mutation rate than nuclear DNA. This fragility contributes to a dozen mitochondrial DNA diseases that range from optic atrophy to Kearns-Sayre syndrome (MIM*530000), not to be confused with similar disorders caused by mutation of nuclear-encoded peptides that are imported into mitochondria. Sperm heads contain few mitochondria, so mitochondrial DNA diseases often exhibit maternal inheritance due to contribution of zygote mitochondria from oocyte cytoplasm. Mitochondrial DNA mutations may arise during oogenesis, causing appearance of a new disease, and may affect only a portion of the cell’s mitochondria, producing mutant-normal mitochondrial mixtures (heteroplasmy). Proportions of mutant mitochondria can differ with age and among tissues, accounting for worsening symptoms, variable ages of onset, and variable symptoms in disorders such as Kearns-Sayre syndrome.
329. The answer is e. (Murray, pp 163-170. Scriver, pp 2261-2274.) Under aerobic conditions, pyruvate is oxidized by pyruvate dehydrogenase to acetyl-CoA, which enters the citric acid cycle. The citric acid cycle generates reducing equivalents in the form of FADH and NADH that are converted to oxygen by the electron transport chain to yield abundant ATP. Under anaerobic conditions such as heavy exercise, pyruvate must be converted to lactate to recycle NADH to NAD+ to allow glycolysis to continue. In mitochondrial disorders resulting from mutations in cytochromes or pyruvate dehydrogenase, there is deficient NADH oxidation and ATP production. Lactate will accumulate as it does normally in tissues without mitochondria (erythrocytes) or in tissues with exercise stress (like muscle). The lactate can accumulate in serum, causing a decreased pyruvate to lactate ratio and lactic acidosis that are typical signs of mitochondrial disease. These abnormalities also occur with circulatory failure (shock) or hypoxemia, so they are suspect for inborn errors only when cardiorespiratory function is normal. Glycolysis produces only 2 ATP compared to the coupling of citric acid intermediates with electron transport that produces 12 ATP per cycle; tissues highly dependent on the respiratory chain (nerves, muscle, and retina) are predominantly affected in mitochondrial disorders—for example, Leigh disease. Suggestive signs such as the decreased pyruvate/lactate ratio must be followed by more specific tests such as muscle biopsy (ragged red fibers), eye examination (retinal pigmentation), or mitochondrial DNA analysis (deletions, point mutations) to diagnose highly variable mitochondrial diseases.
330. The answer is f. (Murray, pp 163-170. Scriver, pp 2327-2356.) The citric acid cycle is closely associated with oxidative-phosphorylation (ox-phos), producing NADH and FADH2 reducing molecules that are reoxidized to NAD+ while generating ATP. The dehydrogenases, isocitrate dehydrogenase, α-ketoglutarate dehydrogenase, and malate dehydrogenase, each produce one NADH, while succinate dehydrogenase produces one molecule of FADH2 per turn of the cycle (incorrect answer e). Glycolysis generates ATP energy under anaerobic conditions when oxygen is not available for coupling with phosphorylation (incorrect answers a, b). Gluconeogenesis consumes ATP/GTP energy to generate glucose necessary for brain and other tissues (incorrect answers c, d). Reoxidation of each NADH results in formation of 3 ATP, and reoxidation of FADH2 results in production of 2 ATP for each molecule of coenzyme A entering the citric acid cycle. Succinate dehydrogenase deficiency (MIM*255125) is a rare muscle disorder (myopathy) that impairs flow through the citric acid cycle, coupling of FADH2 with ATP production, oxidative-phosphorylation, and energy production. Although these processes are sufficient under low-energy conditions, stress in muscle (exercise) with ineffective ox-phos due to succinate dehydrogenase deficiency simulates anaerobic conditions with high lactate, less ATP and energy, less creatinine-phosphate shuttled to muscle cytoplasm, and higher amino acids such as alanine, which usually are converted by transamination to citric acid cycle intermediates, then to oxaloacetate, then to glucose through gluconeogenesis.
331. The answer is c. (Murray, pp 163-170. Scriver, pp 2327-2356.) Oxaloacetate could theoretically stimulate two pathways that may be deficient in the patients (answers a-b, d-e incorrect). Oxaloacetate participates in the first step of the citric acid cycle by accepting an acetyl group from acetyl-CoA to form citrate. Citrate is subsequently metabolized to succinate and back to oxaloacetate, making oxaloacetate a catalyst for the citric acid cycle. Oxaloacetate can also be converted to phosphoenolpyruvate by phosphoenolpyruvate carboxykinase and thus becomes a substrate for production of glucose through gluconeogenesis.
332. The answer is d. (Murray, pp 151-162. Sciver, pp 2327-2356.) In addition to the citric acid cycle that includes conversion of oxaloacetate to citrate, mitochondria also house the enzymes for β-oxidation of fatty acids and oxidative phosphorylation. The cytosol is the site for glycolysis (incorrect answers a, e), glycogenesis (incorrect answer b), the uronic acid and pentose phosphate pathways (incorrect answer c), and fatty acid biosynthesis.
333. The answer is c. (Murray, pp 109-114. Scriver, pp 2261-2296.) Mutations in nuclear or mitochondrial DNA may disrupt oxygen-coupled ATP energy generation (oxidative phosphorylation) in mitochondria and simulate anaerobic conditions. Although exercising muscle fibers can exceed respiratory chain capacity and generate lactic acid through cytosolic glycolysis (incorrect answers a, b), the lactate is quickly converted to glucose by the liver. Cytosolic glycogen breakdown (incorrect answers d, e) does have an energy advantage over glycolysis by yielding glucose 1-phosphate, interchangeable with glucose 6-phosphate, and avoiding the molecule of ATP consumed by hexokinase (which converts glucose to glucose 6-phosphate). When muscle energy deficits become severe, as with mitochondrial, muscle glycogen storage, or even coronary artery diseases with oxygen depletion, then lactate accumulates in cells and serum with muscle cell death, pain, and spasms (cramping).
334. The answer is b. (Murray, pp 109-114. Scriver, pp 2367-2424.) The enzyme creatine phosphokinase (CPK) transfers high-energy phosphate from ATP to creatinine when energy levels are high in resting muscle and ATP is plentiful (incorrect answers a, c-e). The high concentration of CPK in muscle cells causes transfer to serum in various muscle disorders accompanied by muscle cell death (eg, muscular dystrophies with abnormal contractile protein structure, myopathies due to abnormal muscle growth, electrolytes, etc). Elevated serum CPK levels can provide a hint that muscle weakness is due to inherent muscle problems rather than innervation from the brain or peripheral nerves.
Creatine phosphate has a more negative standard free energy of hydrolysis than ATP, whereas ADP, glucose 1-phosphate, and pyrophosphate all have lower energy phosphate groups than ATP. When ATP is utilized rapidly in skeletal muscle, creatine phosphate can be hydrolyzed and act as a phosphate donor to ADP to regenerate ATP. During resting periods when the ATP/ADP ratio is high, creatine can be phosphorylated to creatine phosphate to serve as storage for high-energy phosphate.
335. The answer is c. (Murray, pp 608-628. Scriver, pp 2367-2424.) Muscle contraction is caused by the release of calcium from the sarcoplasmic reticulum following nerve stimulation. In addition to stimulating contraction, the calcium released from the sarcoplasmic reticulum binds to a calmodulin subunit on phosphorylase kinase. This activates phosphorylase kinase, converting it from the D form to the A form (answers a-b, d-e incorrect). The activated phosphorylase then breaks down glycogen and provides glucose for energy metabolism during exercise. In this way, muscle contraction and glucose production from glycogen are coordinated by the transient increase of cytoplasmic calcium levels during muscle contraction.
336. The answer is c. (Murray, pp 207-215. Scriver, pp 2297-2326.) Carnitine acts as a Charon-like molecule that unites with organic and fatty acids as acylcarnitines and transports them across the mitochondrial membrane into the matrix that contains enzymes for fatty acid oxidation. Carnitine deficiency will reduce availability of fatty acids for oxidation and deplete energy to cause fatigue. Fatty acid synthase deficiency would be lethal and reduce muscle fat stores (incorrect answer a), while lipoprotein lipase deficiency would cause serum lipoprotein abnormalities (incorrect answer e), biotin deficiency diverse problems in carboxylation reactions (incorrect answer d), and Tay-Sachs disease neurologic symptoms from accumulation of abnormal brain lipids (incorrect answer b). Carnitine deficiency can result from mutations in a specific carnitine transporter (MIM*212140) or occur in preterm babies with liver problems and dialysis patients. Blockage of the transport of long-chain fatty acids into mitochondria not only deprives the patient of energy production, but also disrupts the structure of the muscle cell with the accumulation of lipid droplets. Oral dietary supplementation usually can effect (bring about?/affect) a cure.
337. The answer is e. (Murray, pp 21-280. Scriver, pp 2513-2570.) Although defects in any of these enzymes will result in a buildup of ammonia in the bloodstream and ammonia intoxication, blocks at carbamoyl phosphate synthase I and ornithine transcarbamylase are usually more severe. Ornithine transcarbamylase deficiency (MIM*300461) is an X-linked recessive disorder, allowing for mild manifestations in female carriers; deficiencies in the other four enzymes are autosomal recessive traits. Gene therapy approaches to treatment are being tested, but resulted in the death of one patient due to suspected reaction to the adenovirus vector.
338. The answer is c. (Murray, pp 548-567. Scriver, pp 3897-3964.) Inability to absorb cobalamin (vitamin B12) from the gastrointestinal tract and its deficiency in vegetarian diets cause megaloblastic anemia and neurologic symptoms (numbness, extremity weakness, poor coordination, and dementia—incorrect answers a, b and d, e). The presence of neurologic symptoms led to the term “pernicious anemia” because they are progressive and eventually irreversible. The absorption of vitamin B12 from the intestine requires a binding protein called intrinsic factor that is secreted by the gastric mucosa and absorbed in the ileum; Pernicious anemia can result from gastric atrophy at older ages or, more rarely, from mutations affecting the intrinsic factor itself that present in childhood (eg, MIM*261000). Inability to absorb vitamin B12 from the gastrointestinal tract causes more severe deficiency than nutritional deprivation in vegetarian diets. Intrinsic factor may also be diminished by autoantibodies in autoimmune diseases such as diabetes mellitus or Graves disease (hyperthyroidism). Clinical signs of pernicious anemia may not appear until 3 to 5 years following the onset of vitamin B12 deficiency and the neurologic signs may occur without obvious anemia.
Pellagra is caused by niacin (vitamin B3) and tryptophan deficiency, leading to photosensitive dermatitis and neurologic symptoms. Scurvy is caused by vitamin C deficiency and is characterized by bleeding gums and bone disease. Rickets is softening and deformation of the bones due to vitamin D deficiency or defects in vitamin D processing. Beriberi (neurologic and/or cardiac symptoms) is caused by thiamine (vitamin B1) deficiency and is common in Asians who eat polished white rice minus the thiamine-rich husk.
339. The answer is e. (Murray, pp 548-567. Scriver, pp 3897-3964.) Vitamin A is a fat-soluble vitamin that can be deficient in combination with thiamine and riboflavin deficiencies in dry climates with food shortages, with other fat-soluble vitamins (D, E, K) in disorders associated with intestinal malabsorption, and in hypothyroidism where there is defective conversion of carotene to vitamin A. Carotenes and carotenoids in plants (yellow corn, carrots, sweet potatoes, leafy vegetables, and green peas) are converted to retinaldehyde in the intestinal mucosa (then to retinol), while retinol is found in animal tissues such as egg yolks, fish oils, butter, liver, and kidney. The first symptoms of vitamin A deficiency are dryness of the eyes (xerophthalmia) with decreased vision in dim light (night blindness), followed by photophobia, corneal irritation, ulceration, and destruction of the eye. Dry skin and rashes also occur. The importance of eggs in people with restricted diets was vividly portrayed in James Clavell’s novel, King Rat, where they were prized for prevention of blindness.
Pyridoxine is present in vegetables, cereals, and fruits; niacin in liver, poultry, and eggs; tetrahydrofolate in milk, uncooked fruits, and vegetables; and riboflavin in milk, eggs, meats, and fruits.
340. The answer is d. (Murray, pp 84-93, 163-170. Scriver, pp 3897-3964.) Lipoic acid is a short-chain fatty acid with two sulfhydryl groups that is a coenzyme for the pyruvate dehydrogenase reaction that converts pyruvate to acetyl-CoA. This reaction also requires the vitamin thiamine and commits pyruvate to the citric acid cycle with generation of NADH, then ATP through mitochondrial oxidative phosphorylation. Mutations affecting the pyruvate dehydrogenase enzyme complex cause Leigh syndrome of lactic acidosis and neurologic disease (MIM*256000), one of many mitochondrial disorders that are treated by supplementing with lipoic acid, thiamine, carnitine, and coenzyme Q. The other choices include glucose 1-phosphate or glucose 6-phosphate that can generate energy through glycolysis (incorrect answers a, b), ornithine and arginine that dispose of ammonia through the urea cycle (incorrect answer c), and creatine, lactate that are generated in muscle (creatine cycles with creatine-phosphate to supplement oxidative phosphorylation in muscle, and lactate also accumulates during exercise when limiting oxygen causes muscle to use glycolysis (incorrect answer e).
341. The answer is a. (Murray, pp 548-567, 467-481. Scriver, pp 3897-3964.) Riboflavin deficiency involves the insidious onset of photophobia, a burning sensation in the eyes, sore mouth (stomatitis) and tongue (glossitis), oily skin with rash (seborrheic dermatitis), and weight loss, confusion, dizziness, headache, and weakness. Retinol deficiency would cause night blindness and dry eyes that could be part of the described disorder, niacin deficiency rash (pellagra) with neurologic symptoms, thiamine deficiency heart failure and neurologic symptoms if acute (beriberi) or more chronic neuritis, pyridoxine deficiency infantile convulsions, or peripheral neuritis (numbness and tingling, more common in slow metabolizers of drugs like isoniazid).
342. The answer is e. (Murray, pp 608-628. Scriver, pp 5493-5524.) The major problem in myasthenia gravis is a marked reduction of acetylcholine receptors on the motor endplate where cranial nerves form a neuromuscular junction with muscles (incorrect answers a-d). In these patients, autoantibodies against the acetylcholine receptors effectively reduce receptor numbers. Normally, acetylcholine molecules released by the nerve terminal bind to receptors on the muscle endplate, resulting in a stimulation of contraction by depolarizing the muscle membrane. The condition is improved with drugs that inhibit acetylcholinesterase.
343. The answer is e. (Lewis, pp 132-151. Scriver, pp 193-202.) Amyotrophic lateral sclerosis (ALS) is not always sporadic (isolated cases—incorrect answer c), not always genetic (incorrect answers a, b), and not present at birth (congenital—incorrect answer d). When counseling families, the physician should realize that lay people often assume that a normal family history excludes the possibility of genetic disease despite the frequent occurrence of autosomal or X-linked recessive mutations.
Many common disorders including both forms of diabetes mellitus, schizophrenia, and most isolated birth defects such as cleft palate exhibit multifactorial determination: multiple genes (polygenic inheritance) plus environmental factors interact to determine susceptibility to disease. Often one or more of these determining genes can sustain mutations that have a major effect on susceptibility as when LDL receptor mutations overwhelm dietary and coagulation factors to cause heart attacks at a young age (familial hypercholesterolemia-MIM*14010). Disorders that exhibit multifactorial determination have a recurrence risk that depends on the number of affected relatives—a first child with birth defect like cleft palate confers a 3% to 5% recurrence risk, a risk that is doubled if a parent and first child are affected. Current research is defining molecular markers that associate with susceptibility to common multifactorial diseases, allowing risk modification in the way that HLA type B27 indicates higher risk for ankylosing spondylitis (MIM*106300—many multifactorial disorders are listed in Online Mendelian Inheritance in Man). The superoxide dismutase gene may be mutated in rare cases of familial (autosomal dominant), early-onset ALS (MIM*105400).
344. The answer is b. (Lewis, pp 70-82, 70-94. Scriver, pp 3-45.) The phenotype refers to individual traits or characteristics and the genotypes to gene combinations that determine them. Genetic loci are positions on chromosomes that contain genes; since all chromosomes are paired in humans except the XY of males, most genes are paired and have structures (now defined as DNA sequences) called alleles. During meiotic segregation, each parental gamete receives one paired gene (one allele) from every genetic locus (except males who receive paired alleles from the small homologous XY short arm regions and single alleles from the nonpaired X or Y. The probability of a parental allele being transmitted to offspring is thus one-half, and the probability of a given genotype appearing in offspring is the joint probability of maternal and paternal allele transmission.
For a paternal Dd versus maternal dd mating, the probability of maternal alleles D or d being transmitted is one-half and the probability of transmission of the paternal allele d is 1. The joint probability for a Dd genotype in offspring is thus one-half for D from mother multiplied by 1 for d from father equals one-half. A similar calculation would apply to the joint probability for the dd genotype, giving probabilities of one-half for Dd and one-half for dd, expected ratios of 1 Dd:1 dd in offspring, or a recurrence risk for father’s deaf phenotype of one-half or 50%. More than 30 genes causing deafness have been characterized in humans, with over 75% exhibiting autosomal recessive inheritance rather than the dominant inheritance implied here.
345. The answer is a. (Scriver, pp 5677-5702. Lewis, pp 63-65, 70-86.) The individual probability for each child developing Huntington disease is 1/2 and the probability that all three children will develop the disease is ½ × ½ × ½ = 1/8 (incorrect answers b-e). Huntington disease exhibits autosomal dominant inheritance, meaning that heterozygotes with one normal and one Huntington allele will develop the disease. The husband most likely inherited a normal allele from his mother and a Huntington allele from his father, making him a heterozygote (homozygous affected individuals with Huntington disease are extremely rare because both parents must be affected). The wife most likely has two normal alleles, since she has no family history and no symptoms of disease (she could theoretically have a Huntington allele that has not yet caused disease but this is unlikely given the incidence of 1 in 15-25,000). Note that the presence of an abnormal allele in the wife could not be discounted if this were an autosomal or X-linked recessive disorder.
346. The answer is c. (Lewis, pp 110-126. Scriver, pp 3-45.) Males always transmit their single X chromosome to their daughters. Therefore, a daughter of a male affected with an X-linked disorder is an obligate carrier for that disorder. When the condition is X-linked recessive, as with most forms of color blindness, the daughter is unlikely to show any phenotypic evidence that she is carrying this abnormal gene. Offspring of female carriers are of four types: (1) female carrier with one normal and one mutant allele, (2) normal female with two normal alleles, (3) affected male with a single mutant allele, and (4) normal male with a single normal allele. The chance of having an affected child is thus one-fourth or 25%. If the obligate carrier female gives birth to a son, the chance of the son being color-blind is 50%.
347. The answer is e. (Lewis, pp 110-126. Scriver, pp 5955-5976.) The common forms of color blindness are X-linked recessive, as indicated by the initial 3 of the McKusick number (MIM*304000). The couple’s daughters will be obligate carriers—that is, carriers implied by the pedigree. Using a lowercase c to represent the recessive color blindness allele, the female is most likely XCXC, while her husband is XcY. The Punnett square below indicates that all daughters will be carriers (XcXC), while sons will be normal (XCY). Note again that loci on the X chromosome cannot be transmitted from father to son, since the son receives the father’s Y chromosome.
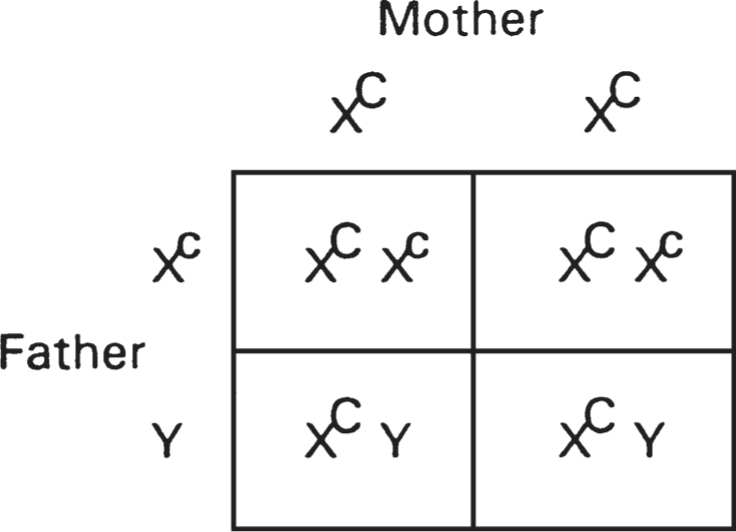
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


