Increasing evidence points to a role for epigenetic changes in human disease in response to environmental or lifestyle influences. The dynamic and reversible nature of epigenetic changes permits a level of adaptability or plasticity that greatly exceeds the capacity of DNA sequence alone and thus is relevant both to the origins and potential treatment of disease. A number of large-scale epigenomics projects (akin to the original Human Genome Project) have been initiated to catalogue DNA methylation sites genome-wide (the so-called methylome), to evaluate CpG landscapes across the genome, to discover new histone variants and modification patterns in various tissues, and to document positioning of nucleosomes around the genome in different cell types, and in samples from both asymptomatic individuals and those with cancer or other diseases. These analyses are part of a broad effort (called the ENCODE Project, for Encyclopedia of DNA Elements) to explore epigenetic patterns in chromatin genome-wide in order to better understand control of gene expression in different tissues or disease states.
DNA Methylation
DNA methylation involves the modification of cytosine bases by methylation of the carbon at the fifth position in the pyrimidine ring (Fig. 3-9). Extensive DNA methylation is a mark of repressed genes and is a widespread mechanism associated with the establishment of specific programs of gene expression during cell differentiation and development. Typically, DNA methylation occurs on the C of CpG dinucleotides (see Fig. 3-8) and inhibits gene expression by recruitment of specific methyl-CpG–binding proteins that, in turn, recruit chromatin-modifying enzymes to silence transcription. The presence of 5-methylcytosine (5-mC) is considered to be a stable epigenetic mark that can be faithfully transmitted through cell division; however, altered methylation states are frequently observed in cancer, with hypomethylation of large genomic segments or with regional hypermethylation (particularly at CpG islands) in others (see Chapter 15).
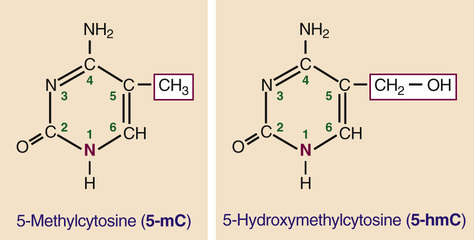
Extensive demethylation occurs during germ cell development and in the early stages of embryonic development, consistent with the need to “re-set” the chromatin environment and restore totipotency or pluripotency of the zygote and of various stem cell populations. Although the details are still incompletely understood, these reprogramming steps appear to involve the enzymatic conversion of 5-mC to 5-hydroxymethylcytosine (5-hmC; see Fig. 3-9), as a likely intermediate in the demethylation of DNA. Overall, 5-mC levels are stable across adult tissues (approximately 5% of all cytosines), whereas 5-hmC levels are much lower and much more variable (0.1% to 1% of all cytosines). Interestingly, although 5-hmC is widespread in the genome, its highest levels are found in known regulatory regions, suggesting a possible role in the regulation of specific promoters and enhancers.
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


