Anaplastic (Undifferentiated) Carcinoma
Yuri E. Nikiforov
Raja R. Seethala
DEFINITION
Anaplastic carcinoma is a highly aggressive malignant thyroid tumor composed of undifferentiated cells that demonstrate immunohistochemical or ultrastructural features of epithelial differentiation. It is derived from follicular epithelial cells but is characteristically devoid of morphologic and immunophenotypic markers of thyroid origin. Another term frequently used to designate this tumor is undifferentiated carcinoma. Other synonyms used in the past include spindle and giant cell carcinoma, sarcomatoid carcinoma, pleomorphic carcinoma, metaplastic carcinoma, and carcinosarcoma.
INCIDENCE AND EPIDEMIOLOGY
Anaplastic carcinoma is a rare type of thyroid cancer comprising 1% to 1.7% of all thyroid malignancies registered in two different United States national databases and 1% to 2% of thyroid cancer cases in many other countries.1,2,3,4 The incidence is higher (up to 8%) in some parts of Europe and in several other countries, particularly in association with iodine-deficiency and endemic goiter areas.5,6,7,8 A higher incidence correlates with low socioeconomic status in some studies, possibly because of a delay of diagnosis of well-differentiated thyroid cancer, a precursor for anaplastic carcinoma.8 Over the past decades, the incidence of anaplastic carcinoma has remained unchanged or even slightly decreased in many countries. In the United States, the incidence showed a 22% decrease between 1973 and 2003 based on the Surveillance, Epidemiology, and End Results (SEER) database.9 This decrease may be attributed to eradication of severe iodine deficiency by iodization of food supplies, earlier detection and treatment of well-differentiated thyroid carcinomas, exclusion of medullary carcinomas and lymphomas, and better recognition of poorly differentiated thyroid carcinoma.10
Anaplastic carcinoma is typically a tumor of older adults; only 13% to 25% of patients are younger than 60 years at diagnosis.11,12,13 The tumors are exceedingly rare before the age of 40, although single cases have been documented.14,15,16 The tumor is more common in females, with a female predilection ranging from 1.5:1 to 2.5:1 in large series of patients.13,17,18,19
ETIOLOGIC FACTORS
Preexisting Thyroid Disease
Preexisting thyroid disease, both malignant and benign, is a major etiologic factor for anaplastic carcinoma.
Preexisting Carcinoma
Differentiated thyroid carcinoma affects the risk of anaplastic carcinoma by serving as a direct precursor lesion. Indeed, it is currently believed that many, if not most, anaplastic carcinomas develop through the process of dedifferentiation of a well-differentiated or poorly differentiated thyroid carcinoma. All major types of welldifferentiated follicular cell-derived carcinomas can be involved. As anaplastic carcinoma is considered to be a follicular cell-derived neoplasm, cases of “anaplastic transformation” in medullary thyroid carcinoma20 should not be considered as part of the same disease spectrum.
The existence of anaplastic transformation is strongly supported by the common occurrence of anaplastic carcinoma in patients with a history of previously treated well-differentiated thyroid cancer and by frequent finding of a differentiated component adjacent to the anaplastic carcinoma area on pathologic examination (Fig. 13.1). A review of series with >50 cases and adequate adequate histologic characterization demonstrates the presence of preexisting or coexisting well-differentiated carcinoma in 23% to 78% of cases.14,17,18,20,21 Papillary carcinoma is the most common coexistent carcinoma and is found in >80% of cases showing both components.21 Follicular carcinoma is far less frequent in series published subsequent to the description of the follicular variant of papillary carcinoma, and both conventional type and oncocytic (Hürthle cell) follicular carcinoma can be found.22 Although possibly underdocumented, anaplastic carcinoma is least frequently associated with poorly differentiated carcinomas. Explanations for the underdocumentation may be due to the historical lack of a uniform definition for poorly differentiated carcinoma (see Chapter 12) and low impact of the finding of poorly differentiated carcinoma in those cases where the anaplastic component is detected.
Some cases of anaplastic carcinoma fail to reveal the areas of well-differentiated or poorly differentiated carcinoma. This may be due to the replacement of a less aggressive component by a more malignant tumor, or due to sampling error, or reflect a possibility that some anaplastic carcinomas develop directly, bypassing a stage of more differentiated carcinoma (Fig. 13.2).
Preexisting Benign Thyroid Disease
A history of long-standing goiter is a well-documented risk factor for anaplastic carcinoma development. In a large cohort of thyroid cancer cases from the United States, ˜25% of patients had a personal history of goiter.13 The prevalence of preexisting goiter was even higher in some other series approaching 50%.20,23 Many patients had a history of goiter present for a very long time, such as for 20 to 60 years.20 It is not entirely clear whether longstanding goiter predisposes to anaplastic carcinoma by increasing the risk of well-differentiated thyroid cancer or independently, by increasing the rate of cell proliferation, or through other and yet unknown mechanisms.
Iodine Deficiency
A higher incidence of anaplastic carcinoma has been reported in areas of dietary iodine deficiency, particularly among those individuals who lived in these areas for at least 20 years or during childhood.7,24 Iodine supplementation in the regions of severe
iodine deficiency coincided with reduction in the incidence of anaplastic carcinoma.25,26 It is likely that iodine deficiency influences the rate of anaplastic carcinoma by increasing the incidence of goiter.
iodine deficiency coincided with reduction in the incidence of anaplastic carcinoma.25,26 It is likely that iodine deficiency influences the rate of anaplastic carcinoma by increasing the incidence of goiter.
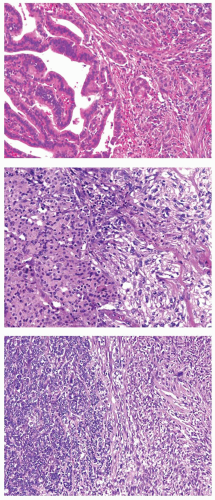 FIGURE 13.1. Anaplastic carcinoma associated with a more differentiated carcinoma component. The latter is a well-differentiated papillary carcinoma (top), well-differentiated oncocytic follicular carcinoma (middle), and poorly differentiated carcinoma (bottom). In each panel, the more differentiated carcinoma component is on the left. |
Radiation Exposure
Almost 10% of patient with anaplastic carcinoma have a history of radiation exposure.13 This suggests the possible role of radiation in tumor development. Studies of large cohorts of patients exposed to ionizing radiation have not found an increase in anaplastic carcinomas, although this may be due to a low overall rate of this tumor occurrence. Ten to twelve percent of patients have a prior history of another malignancy.13,19 This may reflect a general genetic predisposition to carcinogenesis in these patients or be related to the radiation treatment of the first malignancy.
PATHOGENESIS AND MOLECULAR GENETICS
Clonality
Representing the most undifferentiated neoplasm of the thyroid, clonality is not an issue of debate in anaplastic carcinoma. The rare tumors subjected to X-inactivation-based assays confirm this.27 The cytogenetic evolution of multiple “subclones” with different chromosomal alterations has been documented, although this should not be construed as true polyclonality.28
Cytogenetic Abnormalities
Anaplastic carcinoma typically has a complex karyotype with multiple and diverse numerical and structural chromosomal abnormalities.29,30 By comparative genomic hybridization, the mean number of chromosomal alterations ranges from 2.4 to 10.31,32,33 There is a progressive accumulation of chromosomal alterations when comparing well-differentiated carcinomas with poorly differentiated carcinomas and finally anaplastic carcinomas, which is in keeping with the multistep dedifferentiation process.33,34 Anaplastic carcinomas arising in a background of follicular carcinoma appear to have more chromosomal instability reflected by an increased number of alterations as compared with anaplastic carcinomas arising from papillary carcinoma or those without a differentiated component.31 This is not surprising in light of a significantly higher frequency of chromosomal instability observed in follicular carcinomas than in papillary carcinomas (see Chapter 10).
The prevalence of specific chromosomal abnormalities varies from study to study, but the most common alterations include gains of chromosome 1p (up to 33%) and 3p (up to 27%), losses of chromosome 5q (up to 33%), and alterations, either gains or losses of chromosome 8 (up to 44%) and 5p (up to 33%).31,32,33 Decrease in the frequency of gains at 7p and increase in losses at 7q may coincide with anaplastic transformation of follicular and poorly differentiated carcinomas.34
DNA Ploidy
Loss of Heterozygosity
In correlation with multiple large-scale chromosomal alterations detected cytogenetically, loss of heterozygosity (LOH) is frequent in anaplastic carcinomas. As anaplastic transformation represents an “end-stage” of tumor progression for follicular cellderived neoplasms, it can be expected that tumors will harbor multiple deletions at known tumor suppressor gene loci. The mean fractional allelic loss (i.e., frequency of LOH at specific
chromosomal loci) in anaplastic carcinoma is high and ranges from 0.20 to 0.72.37,38,39 Among the most frequent regions of LOH are 1q, 9p, 17p, 16p, 17q, and 18q. When there is a differentiated component, a subset of LOH may be common to both components, although the anaplastic carcinoma component has additional losses, providing molecular evidence for anaplastic transformation of well-differentiated tumors.38,40
chromosomal loci) in anaplastic carcinoma is high and ranges from 0.20 to 0.72.37,38,39 Among the most frequent regions of LOH are 1q, 9p, 17p, 16p, 17q, and 18q. When there is a differentiated component, a subset of LOH may be common to both components, although the anaplastic carcinoma component has additional losses, providing molecular evidence for anaplastic transformation of well-differentiated tumors.38,40
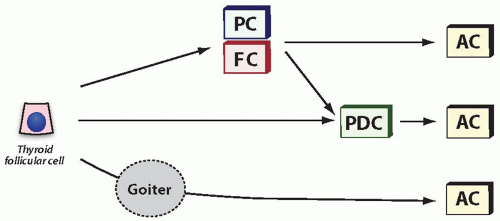 FIGURE 13.2. Putative pathways in the development of anaplastic carcinoma. Current evidence suggests that anaplastic carcinoma may develop by anaplastic transformation of well-differentiated papillary carcinoma well-differentiated follicular carcinoma or poorly differentiated carcinoma or directly from follicular cells, particularly in a setting of a long-standing goiter. AC, anaplastic carcinoma; PC, papillary carcinoma; FC, follicular carcinoma; PDC, poorly differentiated carcinoma. |
Somatic Mutations
Mutations that occur in anaplastic carcinomas can be divided into two groups. One includes genetic alterations that are found in both anaplastic and well-differentiated carcinomas, such as BRAF and RAS mutations. The fact that they are present in welldifferentiated carcinomas suggests that these mutations are early events in thyroid tumorigenesis and are insufficient alone to induce tumor dedifferentiation. Although insufficient for dedifferentiation, they predispose to acquisition of subsequent events that will govern anaplastic transformation. Of note, other mutations commonly found in well-differentiated papillary and follicular carcinomas, such as RET/PTC and PAX8/PPARγ, are rare in anaplastic carcinomas, suggesting that they do not promote tumor dedifferentiation. This is particularly true for RET/PTC1, whereas RET/PTC3 rearrangement has been seen in some papillary carcinomas prone to dedifferentiation.41,42 The other group includes TP53 and β-catenin (CTNNB1) mutations, which frequently occur in anaplastic carcinomas but not in well-differentiated cancers. These mutations are likely to be directly responsible for anaplastic transformation (Fig. 13.3).
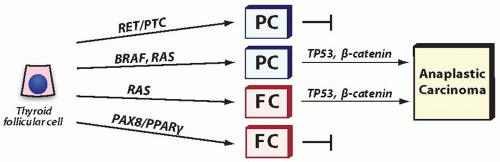 FIGURE 13.3. Gene mutations in the development of anaplastic carcinoma. BRAF and RAS mutations are found in both well-differentiated and anaplastic carcinomas indicating that they occur early and likely to predispose to anaplastic transformation, which is governed by TP53 and β-catenin (CTNNB1) mutations. Tumors with RET/PTC (particularly RET/PTC1) and PAX8/PPARγ are not prone to anaplastic transformation. Mutations in the PIK3CA and PTEN genes are also found in anaplastic carcinomas, although their exact placement in the scheme of stepwise progression is not entirely clear. |
RAS Mutations
Activating point mutations of RAS are common in follicular adenomas, follicular carcinomas, and follicular variant of papillary carcinomas (see Chapters 8, 10, and 11). In anaplastic carcinomas, the prevalence of RAS mutations ranges from 10% to 60% and is ˜30% in the largest reported series.43,44,45,46
As in well-differentiated tumors, the mutations usually affect NRAS codon 61 and HRAS codon 61, although other hot spots (codons 12/13 of NRAS, KRAS, and HRAS) may be occasionally involved. Mutations in codons other than 12, 13, and 61 have been reported, although their functional significance is unclear.47
The differentiated component in these tumors, when present, is typically a follicular carcinoma or follicular variant of papillary carcinoma. An identical RAS mutation is found in both tumor components, consistent with being an early event.
The differentiated component in these tumors, when present, is typically a follicular carcinoma or follicular variant of papillary carcinoma. An identical RAS mutation is found in both tumor components, consistent with being an early event.
The main oncogenic effects of mutant RAS involve the activation of the mitogen-activated protein kinase (MAPK) and PI3K/AKT signaling cascades. Although RAS mutations occur early in tumorigenesis, their frequent presence in anaplastic carcinomas suggests that they are likely to confer the tumors with propensity to dedifferentiate. This may be due to the effect of mutant RAS on promoting chromosome instability and interfering with DNA damage response, which have been demonstrated in cultured thyroid cells.48,49 The increasing instability may predispose the tumor cells to acquire additional mutations, which would in turn initiate anaplastic transformation. The ability of RAS activation to induce the loss of multiple thyroid differentiation markers such as thyroglobulin, thyrotropin-stimulating hormone receptor, sodium/iodide symporter (NIS) and thyroid transcription factor 1 (TTF1) in the cultured thyroid cells50 is likely to be an in vitro phenomenon. Indeed, the presence of RAS mutations in benign thyroid tumors argues strongly against the direct role of RAS activation on the dedifferentiation of human thyroid cells in vivo.
BRAF Mutations
BRAF mutations are common in papillary carcinomas (see Chapter 11) and are found in ˜25% of anaplastic carcinomas.45,51 Many BRAF-positive anaplastic carcinomas contain a well-differentiated papillary carcinoma component, frequently tall cell variant, whereas some tumors show no morphologic evidence of a differentiated component. Similar to papillary carcinomas, the mutations in anaplastic carcinomas are also V600E and are found in both papillary carcinoma and anaplastic carcinoma areas, indicating that they occur early in carcinogenesis.45,52,53
BRAF V600E mutation leads to constitutive activation of the MAPK signaling pathway, and its expression in transgenic mice results in the formation of papillary carcinomas that undergo progression to poorly differentiated carcinomas.54 However, the tumors in these animals did not undergo anaplastic transformation, consistent with the requirement for addition mutations to reach a fully undifferentiated phenotype.
PI3K/PTEN/AKT Pathway Mutations
Mutations in the effectors of this signaling pathway occur in 5% to 10% of well-differentiated follicular carcinomas (see Chapter 10). In anaplastic carcinomas, 6% to 23% of tumors demonstrate PIK3CA mutations and 6% to 16% PTEN mutations.45,46,51,55,56 Most PIK3CA mutations are located at exon 20, the kinase domain, and exon 9, the helical domain, and lead to the activation of this pathway, as demonstrated by the increased phosphorylation of the AKT protein.45 In addition, an increase in PIK3CA gene copy numbers is found in ˜40% of anaplastic carcinomas.51,57 In the PTEN gene, point mutations and small frameshift deletions most frequently occur in exons 5 and 7, leading to the loss of function of the PTEN protein and the subsequent activation of AKT.46
It remains not fully understood whether PIK3CA and PTEN mutations represent an early or late event in thyroid carcinogenesis. They are present with a similar or slightly higher prevalence in anaplastic carcinomas as compared with well-differentiated papillary and follicular carcinomas. However, in contrast to BRAF and RAS mutations, which almost never coexist in the same tumor, PIK3CA and other mutations are frequently seen in anaplastic carcinomas coexisting with BRAF or RAS mutations, suggesting that they may be a late event.45,51 It has also been suggested that gradual increase in AKT stimulation is required for progression from adenoma to well-differentiated follicular carcinoma and then to anaplastic carcinoma.46
TP53 Mutations
TP53 mutations are among the most frequent genetic alterations in anaplastic carcinoma and are typically found in 50% to 80% of tumors.58,59,60,61 The mutational profile is based largely on the screening of exons 5 to 8, where most mutations occur in cancer. The distribution of mutations in thyroid anaplastic carcinomas is similar to that of most other malignancies. The mutation sites are scattered along the evolutionarily conserved domains in these exons. G:C to A:T transitions are the prevalent types of mutation, often in CpG-rich regions, although transversions and small deletions and insertions occur as well.62
TP53 is a nuclear transcription factor that plays an important role in cell cycle regulation, DNA repair and apoptosis, functioning as a tumor suppressor gene. Mutations impair TP53 binding to specific DNA sequences and thus inhibit its transcriptional regulation activity. Mutant TP53 accumulates in the nucleus and manifests immunohistochemically as increased staining (see section Immunohistochemistry). TP53 is commonly altered as a late event in carcinogenesis involving various tumor types. In thyroid cells, TP53 mutations appear to be crucial for the process of tumor dedifferentiation. As such, they are far more common in anaplastic carcinomas than in poorly differentiated carcinomas and are practically absent in well-differentiated carcinomas. In tumors that contain anaplastic and well-differentiated carcinoma components, TP53 mutations are detected only in the anaplastic component.40,63 The best illustration of the multistep tumor progression is provided by a case where a well-differentiated component showed a RAS+/TP53– genotype, whereas the adjacent anaplastic carcinoma was RAS+/TP53+.64
In mouse models, the loss of TP53 in well-differentiated tumors induces anaplastic transformation.65 Reintroduction of wild-type TP53 in anaplastic carcinoma cell lines carrying TP53 mutation results in the reexpression of thyroid specific genes such as TPO and PAX8 and the reacquisition of the ability to respond to thyroid-stimulating hormone stimulation.66,67 This provides another evidence for the role of TP53 in anaplastic transformation and also points to the restoration of TP53 function as a potential therapeutic approach for these tumors.
β-Catenin (CTNNB1) Mutations
Mutations in the gene coding for β-catenin represents another event involved in anaplastic transformation. β-Catenin is a cytoplasmic protein encoded by the CTNNB1 gene and involved in cell adhesion as well as in the wingless (Wnt) signaling pathway.68,69 In the absence of Wnt signaling, β-catenin is expressed mainly on the cell surface, whereas cytoplasmic protein is rapidly degraded after phosphorylation by a multiprotein complex, which includes the adenomatous polyposis coli (APC) protein. Wnt antagonizes this degradation and allows β-catenin to relocate to the nucleus to stimulate target genes. APC and β-catenin mutations can interfere with degradation, resulting in nuclear localization and gene activation promoting tumorigenesis. Point mutations in exon 3 of β-catenin affect the GSK3β phosphorylation sites required for degradation. These mutations are seen in up to 65% of anaplastic carcinomas and with lower prevalence in poorly differentiated thyroid carcinomas.70,71 They are not found in well-differentiated thyroid carcinomas, with the exception of the cribriform-morular variant of papillary carcinoma.72 Unlike this rare variant of papillary carcinoma, β-catenin mutations in anaplastic carcinomas are often multiple, on average 2.4 mutations per case, indicative of the high level of genetic instability.70,71 Mutational status correlates, although not always, with nuclear immunoexpression of β-catenin.
Other Genetic Alterations
Other components of the Wnt signaling pathway may be altered in anaplastic carcinomas. For instance, APC mutations occur in
about 10% of cases. On the basis of limited evidence, it appears that these mutations are mutually exclusive with β-catenin mutations.73 Neither EGFR amplification nor mutations at exons 18, 19, and 21 are present in anaplastic carcinomas, although there is a high frequency (50%) of polysomy for chromosome 7, where EGFR is located.74,75,76 Although loss of TP53 function promotes anaplasia in mice with papillary carcinomas induced by RET/PTC1,65 RET/PTC1 rearrangement is rarely found in human anaplastic carcinoma suggesting that it is less likely to predispose to tumor dedifferentiation than RAS and BRAF mutations.47,77
about 10% of cases. On the basis of limited evidence, it appears that these mutations are mutually exclusive with β-catenin mutations.73 Neither EGFR amplification nor mutations at exons 18, 19, and 21 are present in anaplastic carcinomas, although there is a high frequency (50%) of polysomy for chromosome 7, where EGFR is located.74,75,76 Although loss of TP53 function promotes anaplasia in mice with papillary carcinomas induced by RET/PTC1,65 RET/PTC1 rearrangement is rarely found in human anaplastic carcinoma suggesting that it is less likely to predispose to tumor dedifferentiation than RAS and BRAF mutations.47,77
Profound alterations in gene expression, microRNA, and protein composition are also found in anaplastic carcinomas,78 although the relevance of specific genetic events to pathogenesis of this tumor awaits further analyses.
CLINICAL PRESENTATION AND IMAGING
The classic presentation of anaplastic carcinoma is a rapidly growing neck mass, which is noted in nearly all cases.17 Tumor volume may double as rapidly as in 1 week.79 The mean tumor size at presentation is about 6 cm.19 Other frequent presentation symptoms include hoarseness, dysphagia, and vocal cord paralysis. Up to 50% of patients have extrathyroidal extension at presentation. The most commonly invaded structures include extrinsic muscles, trachea, esophagus, larynx, and laryngeal nerves. Some patients may present with weight loss. Distant metastases are found at presentation in ˜45% of patients (Fig. 13.4), and a similar proportion of patients have cervical lymphadenopathy.13,17,19 The most common sites of metastasis are lungs, bones, and brain. Patients are generally euthyroid on presentation, but rare case reports of transient thyrotoxicosis, presumably secondary to destruction of thyroid parenchyma and rapid release of thyroid hormone, have been described.80,81,82 Some patients may have leukocytosis as a result of secretion of macrophage colony-stimulating factor.83,84 Additional rare clinical presentations include venous thrombosis and humoral hypercalcemia of malignancy, the latter due to parathyroid hormone-related protein secretion.84,85,86
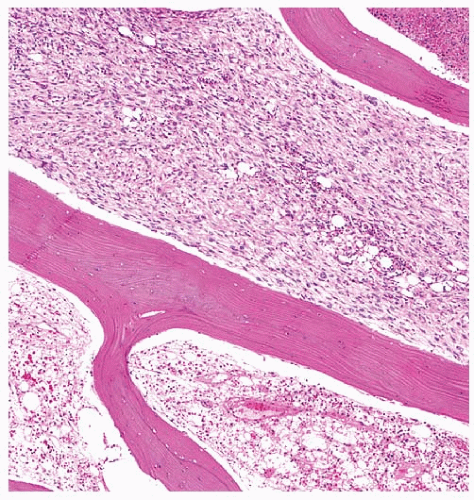 FIGURE 13.4. Anaplastic carcinoma presenting as a distant metastasis to the femur. The tumor is seen on top, permeating between bony trabeculae. |
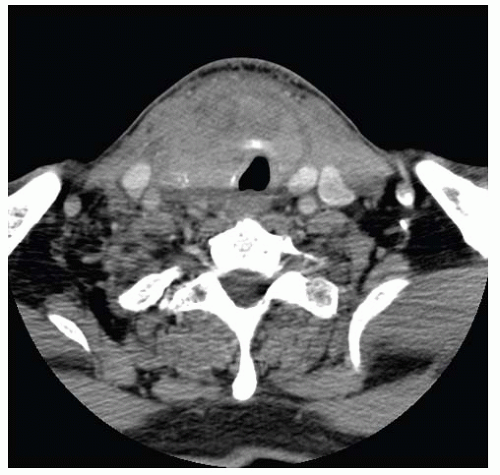 FIGURE 13.5. CT scan of anaplastic carcinoma. It shows an infiltrative heterogeneous mass with thyroid cartilage invasion and encroaching on the adjacent vasculature. |
CT is the preferred method to characterize disease in the neck, whereas MRI is indicated to assess mediastinal extent.87 Using either of these diagnostic modalities, anaplastic is typically an infiltrative heterogeneous mass with irregular borders, and necrosis (Fig. 13.5). On MRI, T1-weighted signal intensities vary from low to high, whereas T2-weighted signal intensities are often high.87 Calcifications are common. Carotid and internal jugular vein involvement is frequently seen. Lymph nodes involved often show rim enhancement on CT.88,89 Anaplastic carcinoma does not typically have uptake of radioactive iodine, either in primary or in metastatic foci and is “cold” on scintigraphy. In fact, areas of radioiodine uptake would suggest a differentiated component.12 The role of combined 18F-fluorodeoxyglucose positron emission tomography and CT for preoperative and postoperative disease assessment is not fully defined, although it has been found to be helpful in some cases.90,91
GROSS FEATURES
On gross examination, anaplastic carcinoma is typically a widely infiltrative mass that replaces the thyroid gland and extends into the adjacent soft tissue or skeletal muscle (Fig. 13.6). Tumors may range from 1 to 20 cm in size but are typically >5 cm.92 Fewer than 10% are confined to the thyroid.19 The cut surface typically demonstrates a friable variegated appearance with multiple foci of hemorrhage and necrosis. Firm or granular tan-brown areas should be sampled for histologic evaluation as they may harbor a residual well-differentiated carcinoma. Very rarely, heterologous elements such as bone or cartilage are grossly identified.93 The paucicellular variant, however, typically shows a homogeneous, firm, tan-white cut surface, mimicking Riedel thyroiditis.
MICROSCOPIC FEATURES
Anaplastic carcinoma may manifest as a multitude of microscopic appearances, often within the same tumor case or even same
slide. However, certain features are common to all tumors. They include (1) widely invasive growth, (2) extensive tumor necrosis, (3) marked nuclear pleomorphism, and (4) high mitotic activity. Indeed, the microscopic appearance of anaplastic carcinoma on low-power magnification is almost invariably infiltrative, both within the thyroid parenchyma and in the adjacent tissues. The invasion of perithyroidal fat tissue and skeletal muscle is common (Fig. 13.7A). The characteristic feature is the permeation or “colonization” of the vessel walls in medium-size veins and arteries (Fig. 13.7B,C). Areas of coagulative necrosis with irregular contours are common (Fig. 13.8A) and may demonstrate tumor cell palisading. All tumor cell types have pleomorphic nuclei with highly irregular nuclear contours and thick coarse chromatin (Fig. 13.8B). The cells are arranged in solid sheets,
with no follicle formation or colloid. The latter can be seen in the entrapped nonneoplastic thyroid cells or well-differentiated carcinoma areas.
slide. However, certain features are common to all tumors. They include (1) widely invasive growth, (2) extensive tumor necrosis, (3) marked nuclear pleomorphism, and (4) high mitotic activity. Indeed, the microscopic appearance of anaplastic carcinoma on low-power magnification is almost invariably infiltrative, both within the thyroid parenchyma and in the adjacent tissues. The invasion of perithyroidal fat tissue and skeletal muscle is common (Fig. 13.7A). The characteristic feature is the permeation or “colonization” of the vessel walls in medium-size veins and arteries (Fig. 13.7B,C). Areas of coagulative necrosis with irregular contours are common (Fig. 13.8A) and may demonstrate tumor cell palisading. All tumor cell types have pleomorphic nuclei with highly irregular nuclear contours and thick coarse chromatin (Fig. 13.8B). The cells are arranged in solid sheets,
with no follicle formation or colloid. The latter can be seen in the entrapped nonneoplastic thyroid cells or well-differentiated carcinoma areas.
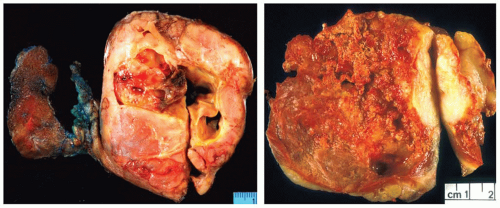 FIGURE 13.6. Gross appearance of anaplastic carcinoma. Left: Tumor replaces the entire right lobe and shows a fleshy, friable cut surface with multiple areas of necrosis and hemorrhage. Right: This tumor infiltrates skeletal muscle (bottom). |
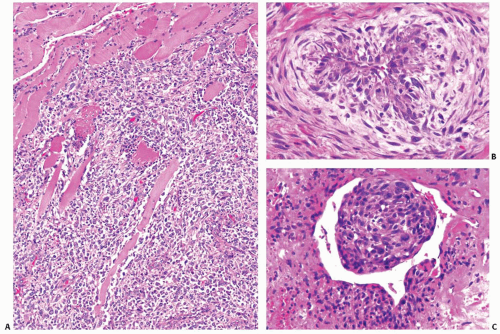 FIGURE 13.7. A: Anaplastic carcinoma infiltrating skeletal muscle. B and C: Anaplastic carcinoma permeating and colonizing a medium-sized artery. The vessel in (C) also shows an intravascular tumor “thrombus.” |
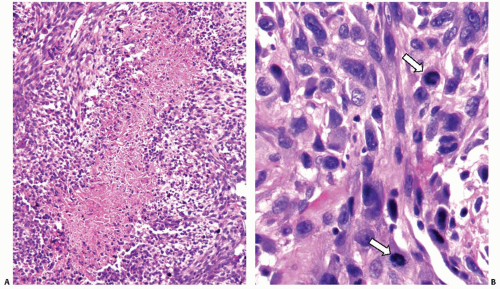 FIGURE 13.8. A: Anaplastic carcinoma demonstrating geographic coagulative necrosis. B: Pronounced nuclear atypia in anaplastic carcinoma. The nuclei show marked pleomorphism, hyperchromasia, and thickened, irregular nuclear membranes. Two mitotic figures are seen (arrows). |
From a cytologic standpoint, anaplastic carcinoma typically contains a mixture of spindled, epithelioid, and pleomorphic giant cells, as described below. Scattered single necrotic cells with pyknotic nuclei are frequently seen. Mitoses are typically abundant and atypical forms may be present (Fig. 13.8B).
The stroma ranges from hyalinized and sclerotic to fibrocellular and desmoplastic. An inflammatory background is common and usually consists of neutrophilic infiltrates. Occasionally (in about 10%), osteoclast-like giant cells may contribute to this inflammatory milieu (Fig. 13.9).12,94 These giant cells contain numerous bland nuclei, which are different from the surrounding highly pleomorphic nuclei of carcinoma cells (Fig. 13.9B,C). They show immunohistochemical and ultrastructural features of monocytic/histiocytic lineage and are likely to derive from histiocytoid mononuclear cells through cellular fusion.95
A preponderance of one cell type gives rise to the common histologic patterns of anaplastic carcinoma.
Spindle Cell Pattern
This is the most common histologic pattern, predominating in about 50% of cases.14,17,79 It recapitulates the appearance of a pleomorphic high-grade sarcoma. Cells may be arranged in a storiform pattern reminiscent of a so-called malignant fibrous histiocytoma, (Fig. 13.10A,B) particularly the inflammatory or myxoid types, or in “herringbone” patterned long fascicles resembling fibrosarcoma (Fig. 13.10C). Occasionally, the spindle cell pattern in anaplastic carcinoma may contain a branching staghorn or “hemangiopericytoma-like” vasculature (Fig. 13.10D). Rarely, hemorrhage and cell dyshesion resulting in an anastomosing network of blood-filled spaces reminiscent of angiosarcoma (Fig. 13.11A).96,97 In <5% of cases, heterologous elements such as malignant bone and cartilage may be seen (Fig. 13.11B,C).9,12
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


