Amyloidosis
A. Brad Farris, III, MD
Key Facts
Terminology
Accumulation of any 1 of > 20 amyloid proteins, characterized by apple-green birefringence after Congo red stain due to β-pleated sheet structure
Clinical Issues
Causes ˜ 5% of nephrotic syndrome in adults
Systemic disease (heart, GI, nerves)
Most due to immunoglobulin light chain (AL amyloid)
AA amyloid due to persistent/recurrent inflammation
Many rare genetic forms
Generally poor outcome
Microscopic Pathology
Amorphous eosinophilic, PAS(+) material in glomerular mesangium and GBM
Defining characteristic: Congo red stain confers apple-green birefringence in polarizing microscope
Also variably present in vessels and interstitium
EM: Straight, nonblanching fibrils, 7-12 nm in diameter
Randomly distributed in mesangium and penetrating GBM
Amyloid protein should be identified by IF/IHC
Light chains (AL amyloid)
AA protein (AA amyloid)
Specific protein in genetic forms
Top Differential Diagnoses
Other diseases with fibrils have no birefringence with Congo red
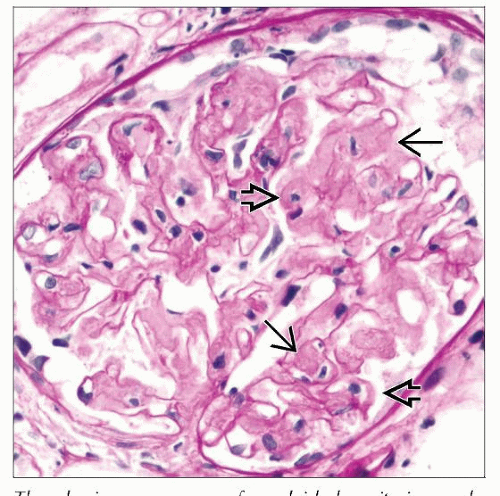 The classic appearance of amyloid deposits is a pale amorphous accumulation in the mesangium  and along the GBM and along the GBM  in PAS stains without cellular proliferation or inflammation. in PAS stains without cellular proliferation or inflammation. |
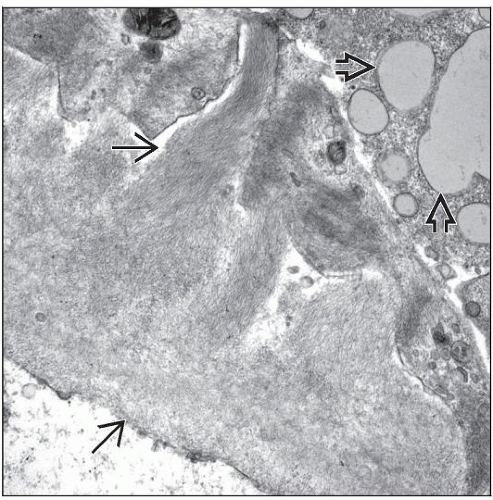 EM reveals subepithelial “spicular” amyloid fibrils  in the GBM. Fibrils are ˜ 7-12 nm in diameter and are nonperiodic. Podocytes have effaced foot processes and reabsorption droplets in the GBM. Fibrils are ˜ 7-12 nm in diameter and are nonperiodic. Podocytes have effaced foot processes and reabsorption droplets  . . |
TERMINOLOGY
Definitions
Protein folding diseases characterized by accumulation of 7-12 nm diameter fibrils with a β-pleated sheet structure that confers birefringence after staining with Congo red
ETIOLOGY/PATHOGENESIS
Amyloidogenic Proteins
> 20 different precursor proteins
Clonal proliferation of plasma cells (AL/AH types)
Chronic inflammation (AA type)
Genetic (multiple proteins)
Failure of excretion (β2-microglobulin)
Neoplasm (e.g., calcitonin)
˜ 90% of renal amyloidosis cases are AL or AA
Pathogenesis
Amyloidogenic proteins have an antiparallel, β-pleated sheet tertiary structure, accounting for Congo red staining and apple-green birefringence under polarized light
Resistance to metabolic processing leads to accumulation and interference with physiologic functioning
Amyloid deposits composed of amyloidogenic protein and nonfibrillary glycoproteins serum amyloid P (SAP), apolipoprotein E, and glycosaminoglycans
SAP (also known as amyloid P component)
Normal plasma protein that binds all amyloid proteins
SAP present early and promotes deposits
Labeled SAP used to image amyloid in vivo
Renal Deposits
Certain forms typically involve the kidney (AL, AA, fibrinogen, Apo AI and AII, Alys)
Glomerular mesangial amyloid accumulation often occurs 1st as mesangial cells lose usual smooth muscle phenotype and acquire macrophage phenotype
Mesangial cells engage in endocytosis and deliver amyloidogenic light chains to lysosomal compartment, where fibril formation takes place
Fibrils penetrate and aggregate in the GBM, suggesting that they may be locally formed
AL Amyloid
Most common cause of renal amyloidosis in the USA/Western hemisphere
Plasma cell dyscrasias or lymphoproliferative disorders
Only ˜ 20% meet diagnostic criteria for multiple myeloma or B-cell leukemia/lymphoma when AL amyloidosis is diagnosed
Some B-cell neoplasms not identified until ≥ 15 years after AL amyloidosis diagnosis
˜ 75% lambda light chains
Often from N-terminal fragment of variable light chain region
40% develop nephrotic syndrome
˜ 10% of AL patients lack glomerular deposits
Formerly called primary amyloidosis
AH Amyloid
Rare cases of AH amyloid composed of monoclonal Ig heavy chains
Can produce nephrotic syndrome
AA Amyloid
Most common cause of amyloidosis in underdeveloped countries
Becoming less common in developed countries
Derived from proteolytic cleavage of serum amyloid A (SAA) protein, an acute phase reactant
Associated with chronic inflammatory conditions
Infections: Tuberculosis, osteomyelitis, bronchiectasis, decubitus ulcers, skin infections of drug abuse
Autoimmune disease: Rheumatoid arthritis, inflammatory bowel disease
Genetic syndromes: Familial Mediterranean fever (FMF), MEFV gene (pyrin)
90% have nephrotic syndrome or renal insufficiency at diagnosis
Formerly called secondary amyloidosis
ATTR (Transthyretin Amyloidosis)
Composed of transthyretin, also known as prealbumin
85% of familial forms of amyloidosis
α-Fibrinogen
Most common genetic form that affects glomerulus
5% of familial forms
β2-Microglobulin
Associated with long-term dialysis
May cause carpal tunnel syndrome, bone cysts, and joint disease
Leukocyte Chemotactic Factor 2 (LECT2)
˜ 2.4% of renal amyloidosis cases
No mutation identified
Other Amyloid Types Affecting Kidney
Cystatin C, gelsolin, apolipoprotein AI or AII, lysozyme
CLINICAL ISSUES
Epidemiology
Incidence
1.4/100,000 per person-year; all types (France)
0.6-1.0/100,000 per person-year; AL type (Minnesota)
About 10% of cases are familial
Age
AL and AA amyloid: Typically 50-70 years old
Familial forms: < 40 years old; AFib older
Gender
M:F = 2:1 overall
Presentation
Proteinuria
Present in virtually all with renal involvement
˜ 5% of adult cases of nephrotic syndrome
Hematuria
Rarely a presenting feature
Extrarenal manifestations
Congestive heart failure, arrhythmias
Dysesthesias
Bladder dysfunction
Orthostatic hypotension
Hepatomegaly/splenomegaly
Macroglossia
Carpal tunnel syndrome
Laboratory Tests
In > 90% of AL amyloidosis patients, monoclonal Ig can be found in blood or urine
Most remaining patients have abnormal monoclonal bone marrow plasma cell populations
Treatment
Treat underlying disease
Myeloma
Alkylating agents (e.g., melphalan) may ↑ survival from 1 year to > 5 years
Bortezomib
Bone marrow transplantation
Inflammatory condition (e.g., immunosuppressive agents for rheumatoid arthritis)
Useful for AA amyloidosis
Colchicine useful for FMF but not as useful for other causes of AA amyloidosis
Transplantation
Recurs post transplant (10-20%); graft failure in 33%
Outcome can be good in selected patients
AL in nonmyeloma patients without severe extrarenal disease
Prognosis
Variable but generally poor
Median survival: 2 years (Mayo series, 859 patients)
MACROSCOPIC FEATURES
General Features
Enlarged, pale, firm, and with a “waxy” appearance
Cut surfaces remain firm and flat in contrast to a normal kidney, which bulges after sectioning
Lugol iodine stains glomeruli dark brown in cut surfaces (like starch)
MICROSCOPIC PATHOLOGY
Histologic Features
Glomeruli
Expansion of mesangium and thickening of capillary walls by amorphous eosinophilic material
Amorphous deposits are acidophilic, “salmon orange”
Usually less acidophilic than collagen (i.e., mesangial sclerosis, interstitial fibrosis)
Normal mesangial matrix, presumably destroyed by activated metalloproteinases, is replaced by amyloid fibrils
Nodular mesangial expansion can often be seen
Amyloid is weakly PAS positive, less than GBM
Silver stains: Expanded mesangial areas but ↓ or no silver staining (i.e., “loss of argyrophilia”)
GBMs also may be engulfed by the material, showing up as areas of complete GBM discontinuity
Subepithelial spikes in capillary loops (“cock’s comb”)
Amyloid deposition can 1st be seen in mesangium and blood vessel walls
Early mesangial deposits may be quite small and subject to being overlooked, resulting in erroneous diagnosis of minimal change disease/glomerulopathy
Little or no hypercellularity
Eventually, ESRD kidneys with glomerulosclerosis from amyloid can be suspected by pale staining on JMS or PAS stains
Mesangial and subendothelial deposits eventually obliterate glomeruli
Rare cases of AL and AA amyloid crescents
Trichrome stains blue in amyloid, usually paler than trichrome staining of collagen
Tubulointerstitium
Congo red staining useful in identifying vascular and interstitial amyloid
Helps distinguish from interstitial fibrosis (IF)
Interstitial involvement often contiguous with involvement of TBMs and vessels
Eventually, IF and tubular atrophy (TA) may be extensive, admixed with interstitial inflammation
Tubular casts containing amyloid are sometimes present and, in rare case reports, are the only manifestation of amyloidosis
Material has fibrillar appearance characteristic of amyloid on EM
Interstitium may be expanded by amyloid material
Mast cells may lead to IF in AA type
Blood vessels
Any vessel can be involved, including vasa recta of renal medulla
Blood vessel amyloid can look like hyalinosis
Congo red staining helps distinguish amyloid deposition from hyalinosis
Interlobular arteries and hilar arterioles are most commonly involved (> 95% of renal biopsies having glomerular involvement)
ANCILLARY TESTS
Immunofluorescence
Pattern depends on type of amyloid
Deposits variably present in glomerular mesangium, GBM, interstitium, and vessels
AL: Single light chain predominates
AA: Neither or both light chains stain
Fibrinogen (Aα chain) only glomerular
Kappa and lambda staining performed on all native renal biopsies, identifying most cases of AL amyloidosis
Positive stain is when 1 light chain clearly predominates
Light chains may be present in other forms of amyloidosis due to nonspecific trapping
Negative stains do not exclude AL amyloid
Staining may be negative if light chain is truncated
Variants
Rare cases stain for an immunoglobulin heavy chain but not for light chains (AH amyloidosis)
Rare cases have 1 heavy chain (typically gamma) and 1 light chain (AL/AH amyloid)
Electron Microscopy
Fibrils
Nonbranching, nonperiodic, ˜ 7-12 nm in diameter
Accurate measurement necessary
Internal references: Cell membrane 8.5 nm; actin: 5 nm
Amorphous “cottony” appearance at ˜ 5,000x
Electron-lucent core at ˜ 100,000x
Randomly distributed in mesangium and GBM
Fibrils sometimes extend transmurally, replacing entire glomerular basement membrane
In subepithelial zone, fibrils align roughly perpendicular to GBM, producing “spike” formation or “cock’s comb” (also called “spicular amyloid”)
Podocyte foot processes often effaced, and cytoplasm has condensation of actin filaments
Detection of Amyloid
Congo red
Apple-green birefringence with polarized light
In contrast to fibrillar collagen, amyloid fibrils are only birefringent after Congo red stain
Important to have simultaneous positive control
Red without polarization (called “congophilia”)
Elastic fibers are congophilic but not birefringent
Small amyloid quantities may make it difficult to demonstrate apple-green birefringence
Higher quality polarizing microscope will have greater sensitivity
9 µm sections recommended for Congo red staining to maximize detection of small quantities of amyloid
Placing Congo red stains under green fluorescent light makes amyloid deposits appear bright red; sensitive but not specific
Potassium permanganate treatment prior to Congo red staining used to discriminate between AL and AA amyloid
Birefringent Congo red staining retained by AL amyloid but lost by AA
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


