Chapter 26 Amino Acid Metabolism
Amino acids are used for three major purposes:
Amino Acids Can Be Used for Gluconeogenesis and Ketogenesis
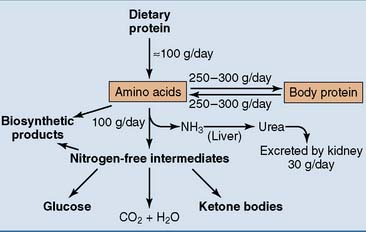
Figure 26.1 shows an overview of amino acid and protein metabolism. A typical dietary protein intake is 100 g/day. Another 250 to 300 g of amino acids comes from protein breakdown, but the same amount is consumed for protein synthesis. Different proteins turn over at different rates. Most metabolic enzymes have a life expectancy of one or a few days, but most plasma proteins circulate for 1 to 3 weeks. Hemoglobin survives for 120 days, collagen lasts up to several years in some tissues, and the lens proteins last for a lifetime.
The Nitrogen Balance Indicates The Net Rate of Protein Synthesis
The nitrogen balance is the difference between the nitrogen entering the body and that leaving it. A normal adult with adequate protein intake should be in nitrogen equilibrium (Fig. 26.2). This means that the amount of outgoing nitrogen matches exactly the amount of incoming nitrogen, and the amount of body protein remains constant.
The Amino Group of Amino Acids Is Released as Ammonia
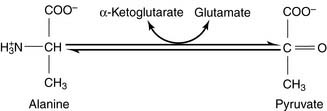
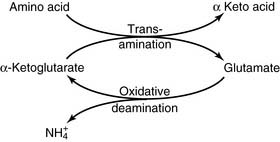
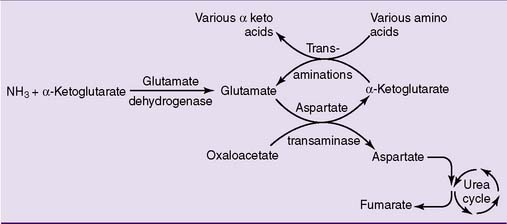
During amino acid catabolism, some of the amino acid nitrogen is released as ammonia. The most important ammonia-forming reaction is the oxidative deamination of glutamate by glutamate dehydrogenase in liver and other tissues (Fig. 26.3, A). This reversible reaction can function in both the synthesis and the degradation of glutamate. The enzyme uses mainly nicotinamide adenine dinucleotide (NAD+) for glutamate degradation and reduced nicotinamide adenine dinucleotide phosphate (NADPH) for glutamate synthesis. The glutamate dehydrogenase reaction implies that glutamate is both nonessential and glucogenic.
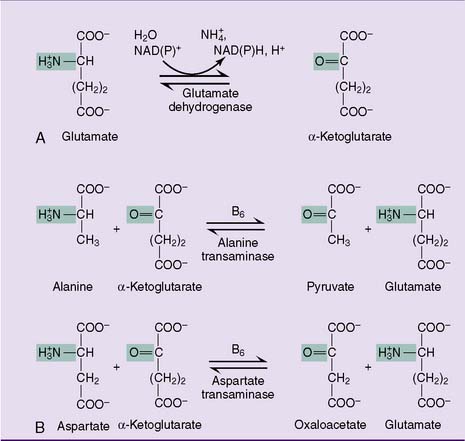
Most other amino acids do not form ammonia directly. They transfer their α-amino group to α-ketoglutarate to form glutamate. The enzymes that catalyze these reversible amino group transfers are called transaminases or aminotransferases. Examples are shown in Figure 26.3, B.
Alternatively, the glutamate that accumulates from these transaminase reactions can be used to make aspartate via aspartate transaminase operating in the reverse direction (Fig. 26.3, B). Both ammonia and aspartate are sources of nitrogen for the synthesis of urea (see section about urea cycle).
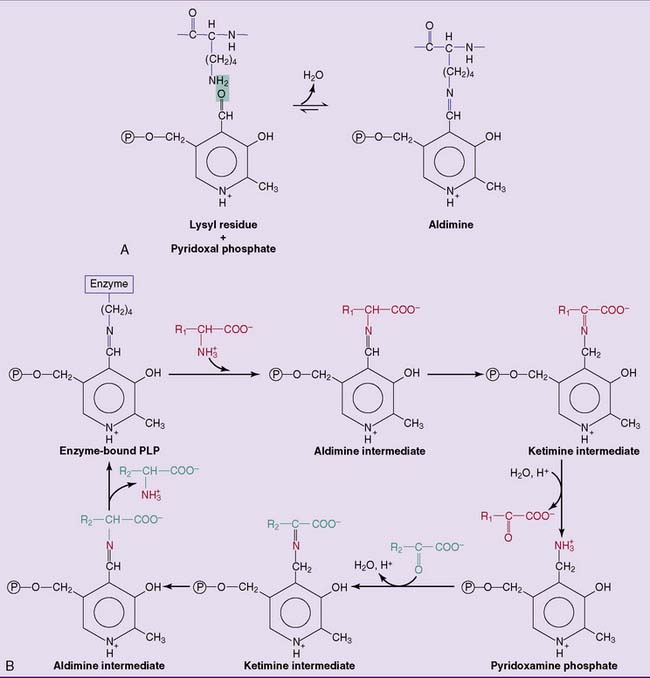
All transaminases contain pyridoxal phosphate (PLP), the coenzyme form of vitamin B6, as a prosthetic group. PLP is bound to the active site of the enzyme by electrostatic interactions and by a Schiff base (aldimine) bond with a lysine side chain of the apoprotein. PLP participates directly in the reaction as shown in Figure 26.4.
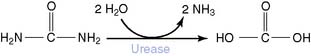
Urea Is Synthesized in the Urea Cycle
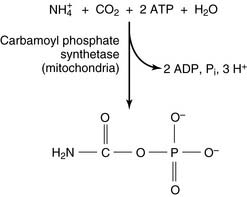
The reactions of the urea cycle proper are shown in Figure 26.5. One of the two nitrogen atoms in urea comes from ammonia via carbamoyl phosphate and the other from aspartate. Most of the aspartate nitrogen is derived from transamination reactions in the liver (Fig. 26.6).
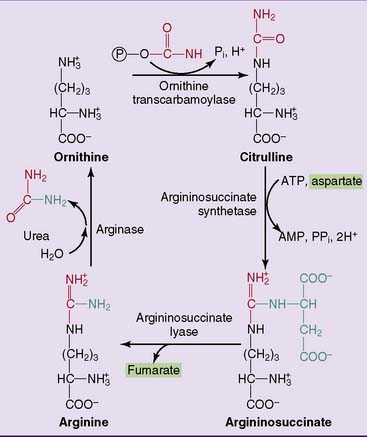
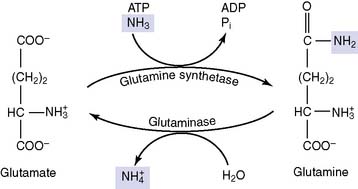
Failure of the urea cycle leads to ammonia toxicity and encephalopathy. Inherited urea cycle enzyme deficiencies (see Clinical Example 26.1) are rare conditions with a combined incidence of 1:8000. In addition to ammonia, glutamine is elevated because excess ammonia is diverted into glutamine synthesis. The substrate of the defective enzyme also accumulates. Death in infancy or severe disability is inevitable when a urea cycle enzyme deficiency is complete, but partial enzyme deficiencies lead to milder impairments.
A far more common cause of hyperammonemia is liver failure. Patients develop a form of encephalopathy that is caused in large part by ammonia toxicity but also is related to other aspects of impaired liver function (Clinical Example 26.2).
CLINICAL EXAMPLE 26.1: Ornithine Transcarbamoylase Deficiency
The diagnosis is established by findings of elevated blood ammonia, glutamine, and ornithine and reduced blood urea nitrogen. Orotic acid appears in blood and urine because accumulating carbamoyl phosphate leaks from the mitochondria into the cytoplasm, where it is a precursor of orotic acid in the pathway of pyrimidine synthesis (see Chapter 28).
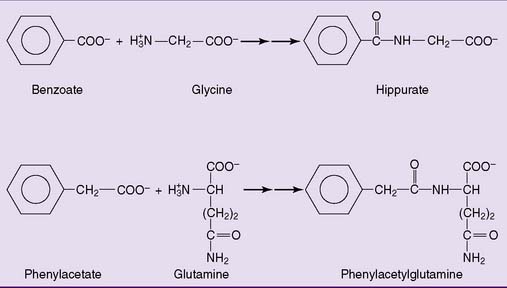
Treatment includes dietary protein restriction, combined with sufficient carbohydrate to prevent the breakdown of body protein for gluconeogenesis. Patients are fed high doses of benzoic acid and phenylacetic acid, which are excreted in the urine as conjugation products with glycine and glutamine, respectively (Fig. 26.7). Ordinarily, these reactions are detoxification reactions that convert the unwanted acids into water-soluble excretable products. In this context, however, they are exploited as alternative routes of nitrogen excretion. Arginine or, preferably, citrulline is administered because the urea cycle functions as a biosynthetic pathway for arginine. For patients with urea cycle enzyme deficiencies, arginine is an essential amino acid.