Alport Syndrome
Helen Liapis, MD
Joseph Gaut, MD, PhD
Key Facts
Etiology/Pathogenesis
85% X-linked inheritance of mutations in COL4A5
15% autosomal recessive COL4A3, COL4A4
Clinical Issues
Presentation
Hematuria, proteinuria, sensorineural deafness, hypertension, eye abnormalities
90% of X-linked males and 12% of X-linked carrier females develop ESRD by age 40
Microscopic Pathology
Early: Minimal glomerular changes, mesangial hypercellularity, interstitial foamy macrophages
Late: Thick capillary loops, FSGS, global sclerosis, tubular atrophy, interstitial fibrosis
Electron microscopy
GBM multilamellation, microparticles, scalloping, and thin GBM
Immunofluorescence: Decreased α3, α4, α5 (IV) collagen in GBM
X-linked male AS: Absent staining
X-linked female heterozygotes: Mosaic loss
Autosomal recessive: α5 (IV) present in BC
Ancillary Tests
Skin biopsy
X-linked form has absent α5 (IV) collagen staining in men, mosaic in women
Diagnostic Checklist
Lamination of GBM can also be seen in repair in other diseases
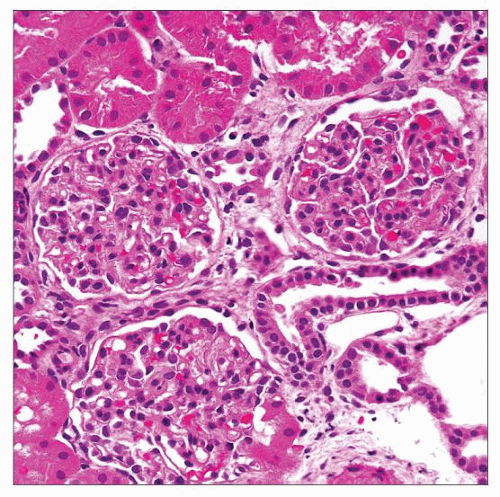 This kidney biopsy is from a 2-year-old boy who presented with proteinuria and a family history of X-linked Alport syndrome. The glomeruli show mild mesangial hypercellularity but are generally unremarkable. |
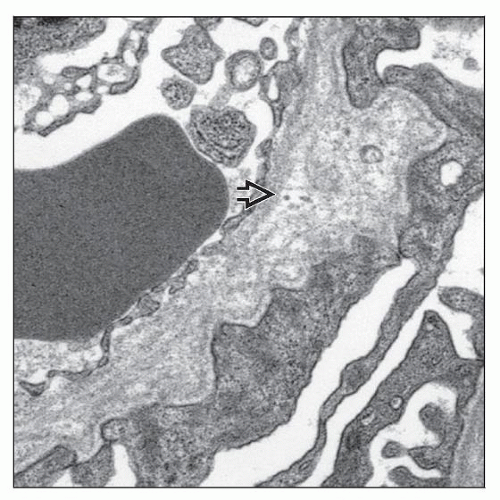 This GBM segment in a boy with X-linked AS shows classic splitting and multilamellation (“basket weaving”), diagnostic of AS. The GBM also contains microparticles (“bread crumbs”)  . . |
TERMINOLOGY
Abbreviations
Alport syndrome (AS)
Synonyms
Hereditary nephritis
Definitions
Inherited glomerular disease secondary to mutations in the α3, α4, or α5 chains of type IV collagen, characterized by hematuria with hearing and eye abnormalities
ETIOLOGY/PATHOGENESIS
Genetics
COL4A5
Encodes α5 type IV collagen chain
X-linked form
Maps to chromosome Xq26-48
85% of patients
10-15% are de novo mutations
Female carriers may show disease depending on degree of mosaicism following lyonization
Mutations in adjacent COL4A6 gene result in diffuse leiomyomatosis
COL4A3
Encodes α3 type IV collagen chain
Autosomal recessive inheritance
Maps to chromosome 2q35-37
Autosomal dominant inheritance has been described but is rare
Disease may result from compound heterozygous or homozygous mutations
COL4A4
Encodes α4 type IV collagen chain
Autosomal recessive inheritance
Maps to chromosome 2q35-37
Heterozygote phenotype in thin basement membrane disease
Autosomal dominant inheritance has been described but is rare
Disease may result from compound heterozygous or homozygous mutations
Mutations of COL4A3 and COL4A4 account for 15% of AS patients
Pathogenesis
Normal GBM and distal TBM composed primarily of α3α4α5 trimeric collagen IV molecules
Deficiency in any 1 of the 3 chains leads to failure of formation of trimer and lack of other 2 chains
Bowman capsule contains α5α5α6 trimers, so expression of α5 not dependent on α3 or α4
α1α1α2 collagen IV chains, minimally present in subendothelial side of normal GBM, increased in AS
Mutations may result in protein misfolding, truncation, or absence of chain
Protein misfolding may lead to degradation of α3α4α5 type IV collagen
Nature of mutation in X-linked form influences age of onset of ESRD
Earliest onset (mean: 25 years) with truncating mutation
Intermediate onset (mean: 28 years) with splice site mutations
Later onset (mean: 37 years) with missense mutations
Mutation at 5′ end with earlier onset and more extrarenal manifestations
CLINICAL ISSUES
Epidemiology
Presentation
Hematuria
Males typically present with gross hematuria
Females typically present with microscopic hematuria
Tends to be persistent in males and intermittent in females
Exacerbated by exercise, infection
Proteinuria, 1-2 g/d
Tends to develop later in disease course
Variable in X-linked AS
Common in autosomal recessive AS
Nephrotic syndrome in 30%
Sensorineural deafness
90% of X-linked hemizygotes by age 40
10% of X-linked heterozygotes by age 40
67% of AR homozygotes before age 20
Hypertension
Eye abnormalities
Anterior lenticonus in ˜ 22% of patients < 25 years old
Pathognomonic of AS
Associated with rapid ESRD and hearing loss
Retinal flecks in ˜ 37% of patients < 25 years old
Leiomyomatosis (rare)
Mutations in COL4A6 and COL4A5
Laboratory Tests
Direct DNA sequencing or linkage analysis of COL4A3/A4/A5
Sensitivity of linkage analysis reported to be ˜ 60%
Treatment
None available to reverse
Transplantation for ESRD
Rarely, anti-GBM disease may develop post transplant
Prognosis
X-linked males
90% develop ESRD by age 40
X-linked carrier females
12% develop ESRD by age 40
60% develop ESRD by age 60
Autosomal recessive
Earlier and more rapid progression to ESRD
Autosomal dominant
Slower progression to ESRD
MICROSCOPIC PATHOLOGY
Histologic Features
Glomeruli
Early changes
Minimal changes
Mild mesangial hypercellularity
Small capillary diameter
Lamination of GBM hard to appreciate by light microscopy (LM)
Late changes
Focal segmental glomerulosclerosis (FSGS)
Global glomerulosclerosis
Interstitium and tubules
Interstitial fibrosis and tubular atrophy
Interstitial foamy macrophages
Vessels
Arteriosclerosis
ANCILLARY TESTS
Immunofluorescence
Electron Microscopy
Transmission
Multilamellation of the GBM lamina densa imparting a “basket weave” appearance
GBM microparticles or “bread crumbs” between laminations
Scalloping or “outpouching” of subepithelial surface of GBM
Irregular, variable GBM thickness, both thick and thin
Podocyte foot process effacement
Thin GBM measuring < 200 nm as only lesion
X-linked carrier females
Autosomal recessive carriers
Identical to thin basement membrane disease
Typically have milder clinical course
Demonstration of Collagen IV α Chains
Normal distribution
α5 is present in GBM, Bowman capsule (BC), distal TBM, collecting duct, and EBM of skin
α3 and α4 are normally expressed in GBM and basement membrane of distal tubules
α1 is abundant in GBM during development; decreases with normal GBM maturation
X-linked AS: Male
Absent α5(IV) staining in GBM, TBM, BC
Absent α3(IV) staining GBM, TBM
α1(IV) increased in GBM
X-linked AS: Female (heterozygote)
α5 and α3 expression may be preserved, decreased, or may show mosaic pattern in GBM and TBM
Autosomal recessive AS
Absent or severely decreased α3 and α5 staining in GBM and distal TBM
Preserved α5 staining in BC
Skin biopsy
EBM shows absent α5(IV) in males with X-linked AS
EBM shows mosaic α5(IV) staining in X-linked female heterozygotes
Normal α5(IV) in autosomal recessive AS
DIFFERENTIAL DIAGNOSIS
Thin Basement Membrane Disease
Normal collagen IV α3-5 staining pattern
Generally no structural damage to glomeruli
IgA Nephropathy
Mesangial IgA deposits
May have laminations of GBM as part of repair
Nail-Patella Syndrome
Type III collagen deposition in GBM by EM
Highlighted using phosphotungstic acid
Lamina densa electron-lucent areas on EM
DIAGNOSTIC CHECKLIST
Clinically Relevant Pathologic Features
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


