New Terminology82
Protein Allosterism82
Types of Allosteric Modulators84
Unique Effects of Allosteric Modulators87
Detecting Allosterism93
Quantifying Allosteric Effect98
Descriptive Pharmacology: V101
Summary102
This chapter begins with a discussion of the characteristic properties of an allosteric molecule, namely saturability of effect (the allosteric modulation ends when the allosteric site is fully occupied), probe dependence (the allosteric effect of a given modulator can be different for different co-binding ligands) and the production of separate effects on the affinity and efficacy of the co-binding ligand. In addition, the strategies for identifying allosterism and the reasons why this is relevant to drug discovery are featured. Specifically, these involve the unique therapeutic properties of allosteric modulators. Finally, the use pharmacologic tools to quantify allosteric behavior of molecules in studies aimed at optimization of allosteric activity will be highlighted. Once allosterism has been identified as a mechanism, then comparison of data to an allosteric model can be used to identify characteristic parameters of the effect.
Keywords
co-operativity, modulator (allosteric), negative allosteric modulator (NAM), positive allosteric modulator (PAM), probe dependence.
Introduction
Conventional concepts about drug discovery center on the assumption that a drug must have an affinity for a naturally physiologically relevant site on a biological target. For instance, antimuscarinic antagonists such as scopolamine are targeted toward the acetylcholine binding site on the acetylcholine receptor. Similarly, many kinase inhibitors are targeted toward the natural ATP binding site of kinases. These systems, as discussed in Chapter 4, can be thought of as “orthosteric” in nature since steric hindrance may play an important role in the mechanism of action. Allosteric molecules can bind to virtually any site on the target protein and affect its activity; this greatly expands the possible therapeutic opportunities for a given target. This may be an extremely important approach, as it is known that if a molecule is bound to the protein at any site the conformational movement of that protein may be affected (vide infra). These effects are allosteric and have become a very important part of new drug discovery.
New Terminology
The following new terms will be introduced in this chapter:
• α: Denoted within the standard model for functional allosterism quantifying the effect of an allosteric modulator on the affinity of a protein for another molecule.
• β: Denoted within the standard model for functional allosterism quantifying the effect of an allosteric modulator on the efficacy of an agonist binding to a receptor protein.
• Co-operativity: The effective interaction between the two co-binding allosteric molecules on the protein, i.e., the effect of one of the ligands on the affinity and efficacy of the other.
• Ensemble: A collection of protein conformations visualized as a snapshot in time in a dynamic system whereby the protein spontaneously samples an enormously large library of conformations.
• Modulator (allosteric): A molecule that co-binds with another on a protein to affect the behavior of the protein toward the cell and the co-binding ligand.
• Negative allosteric modulator (NAM): An allosteric modulator that antagonizes agonist activation of a receptor, i.e., reduces the affinity and/or the efficacy of an agonist for a receptor (an allosteric antagonist).
• Positive allosteric modulator (PAM): An allosteric modulator that promotes agonist activation of a receptor, i.e., increases the affinity and/or the efficacy of an agonist for a receptor.
• Probe dependence: Variation of activity of an allosteric modulator as it modifies the protein interaction with various probes (radioligands, agonists).
Protein Allosterism
Allosteric interactions on proteins such as receptors and ion channels occur through the binding of a molecule onto the protein to affect its free energy of conformation. This subsequently affects the behavior of the protein toward the cell, other proteins and ligands. The change in behavior occurs through a change in conformation of the protein. The energy of the protein changes upon ligand binding no matter how relatively small the molecule is in relation to the protein. There are a number of proposed mechanisms for protein allostery, ranging from the existence of low energy pathways between binding sites (allosteric “hot wires”) to the stabilization of global protein conformations. A major milestone in the consideration of protein behavior was given by Koshland[1] who described how structured enzymes (modeled by the historical “lock-and-key” model of rigid proteins) accommodated substrates through movement of amino acid residues (“induced fit;” see Box 5.1). For receptors, this induced fit model is less applicable than the conformational selection model discussed in Chapter 2 (see Fig 2.6 and Box 2.5) describing the stabilization of global protein conformations through ligand binding. Receptors and other protein drug targets are complex macromolecules that exist in a multitude of tertiary interconvertable conformations (shapes). At any one instant, a collection of protein molecules will have a selection of different conformations of similar free energy; these collections are called ensembles. As discussed in Chapter 2, the binding of a ligand initiates a process of conformational selection within the ensemble where the ligand preferentially binds to the conformations for which it has the highest affinity at the expense of others. If the enriched protein species mediates cellular response then the ligand is an agonist. In this chapter, ligands that produce a conformational change to affect the behavior of the protein toward other molecules will be considered; these molecules are referred to as allosteric modulators.
Box 5.1
In 1894, Emil Fischer accounted for the extraordinary specificity of enzyme–substrate interactions with a description of a strict geometric complementarity; this idea became famous as a “lock-and-key” hypothesis for recognition between molecules and proteins. While this explained enzyme recognition of substrates, it did not account for the stabilization of a new state of the enzyme referred to as the “transition state” needed to induce enzyme catalysis. Daniel Koshland, working at the University of California, Berkely, recognized that proteins are flexible structures and hypothesized that they accommodate the substrate by molding their shape around it; the substrate may also change its shape to optimize a conformation ideal for catalysis. This idea, referred to as “induced fit,” overturned a 100-year-old view of how enzymes function and paved the way for allosteric theories of enzyme and receptor mechanisms.
 |

Protein allostery describes the process of co-operativity between binding sites, i.e., the binding of a molecule at one site on the protein alters the subsequent interaction of the protein with other molecules binding at other sites. One of the earliest models of allosterism in proteins, termed the Monod–Wyman–Changeux model, [2] describes the binding of molecules to proteins made up of multiple subunits, whereby the binding of a molecule to one subunit alters the subsequent binding of molecules to other subunits. Figure 5.1 shows the binding of a molecule to a single subunit and to a trimeric protein consisting of three subunits where the binding of each promotes the binding of the next (positive co-operativity). This produces a steep curve with a slope greater than unity for a Hill coefficient. Such binding behavior can be advantageous in physiology; for example, tetrameric co-operativity of oxygen binding to hemoglobin optimizes the delivery of oxygen to tissues (see Box 5.2).
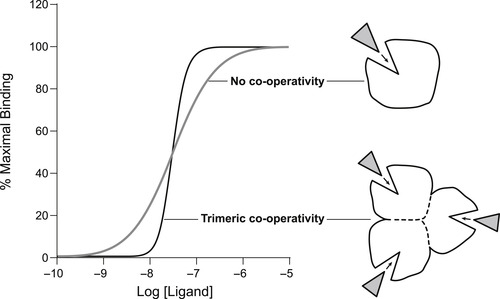 |
| Figure 5.1 Dose–response curve for the binding of a ligand to a single subunit protein according to the Langmuir isotherm (equation 2.1, curve in gray marked “no co-operativity”) and to a protein made up of three identical co-operatively linked subunits (black curve labeled “trimeric co-operativity”). For the trimer the binding of the ligand to one subunit promotes the binding to the second and the third, causing the binding curve to be steeper than that for a monomer. |
Box 5.2
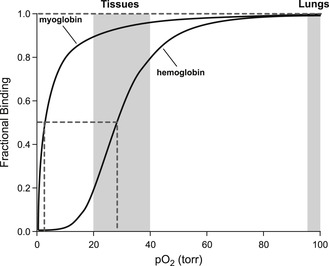
Hemoglobin is a tetrameric protein that binds and transports four oxygen molecules per unit and then releases them to myoglobin. The binding of oxygen to hemoglobin is allosterically co-operative, in that the binding of each oxygen molecule facilitates the binding of the next. As shown on the curve to the right, this produces a steep binding curve ideal for oxygen binding, transport and release. It can be seen from these curves that oxygen readily binds to hemoglobin at the high pO2 values in the lung (100torr). However, in tissues with lower oxygen levels (pO2 20–40torr) the co-operative binding of oxygen to hemoglobin causes the oxygen binding to drop off sharply. This allows hemoglobin to release the oxygen to myoglobin. It can be seen that the non-co-operative binding of oxygen to myoglobin causes it to be more tightly bound in this region of oxygen tension (20–40torr).
The co-operative binding of oxygen is caused by the interaction of the four subunits of hemoglobin, whereby the binding of an oxygen molecule to one subunit increases the affinity of the remaining subunits for oxygen.
 |
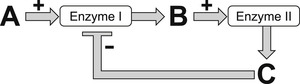
Allosteric ligands bind to their own site on the protein. This opens the possibility of also binding the endogenous ligand as a co-binding ligand. This geographical distinction between binding sites was one of the first features of allosterism to be discovered. Specifically, the modification of biosynthetic enzyme activity, through binding of a structurally unrelated downstream product of biosynthetic pathways (as in the case of the biosynthesis of isoleucine[3]), was one of the first indications that proteins such as enzymes can bind molecules at multiple sites to modify their activity (Fig. 5.2). The binding of molecules to separate sites leads to a permissive system (whereby the action of one of the ligands may not necessarily preclude the action of another); this can facilitate an interaction between the endogenous ligand and the modulator. It also means that the behavior of the modulator-bound protein can vary for different co-binding ligands. This is one of the most important features of allostery, namely probe dependence. If the endogenous ligand is thought of as a probe of receptor function, then a given allosteric modulator can have different effects on different probes. The second important feature of allostery is saturation of effect. Unlike competitive mechanisms where effects can continue as long as different quantities of the interactants are added to the system, allosteric effects cease when the allosteric site on the protein is saturated. These properties of probe dependence and saturation of effect will surface repeatedly throughout this chapter as the specific properties of allosteric modulators are discussed.
Types of Allosteric Modulators
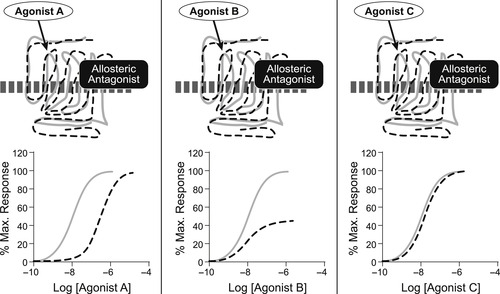
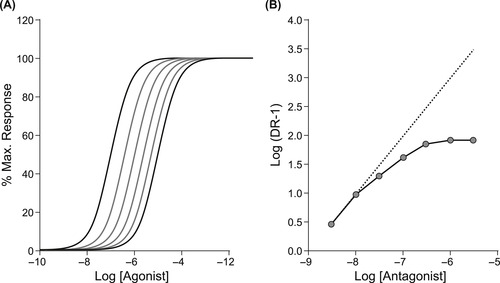
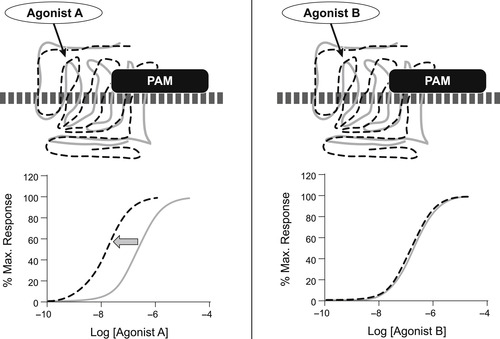
There are no a priori rules for how a given allosteric modulator will affect the action of receptor probes. If the responsiveness of the receptor is reduced to a given probe, then the molecule is an allosteric antagonist (see Fig. 5.3). It should be noted that the principle of probe dependence dictates that different probes may be affected in different ways. Thus, an allosteric antagonist may block some agonists but not others (Fig. 5.3). The pattern of antagonism can vary from being surmountable to being insurmountable. A discerning feature of allosteric antagonists is that they can produce a maximal asymptotic effect (when the allosteric site is fully occupied); the maximal effect they have on a receptor system is determined by co-operativity factors (vide infra). This fact can be used to differentiate allosteric antagonism from orthosteric antagonism. For example, a given surmountable allosteric effect may produce shifts to the right of the agonist dose–response curve that come to a maximal value (see Fig. 5.4A). Under these circumstances, Schild analysis in such a system produces a distinct curvature of the Schild plot (Fig. 5.4B). The use of this concept to identify allosterism is discussed later in this chapter. Some allosteric modulators can also increase the responsiveness of protein targets; these are referred to as positive allosteric modulators (PAMs) (see Fig. 5.5). The effects of PAMS are also subject to probe dependence.
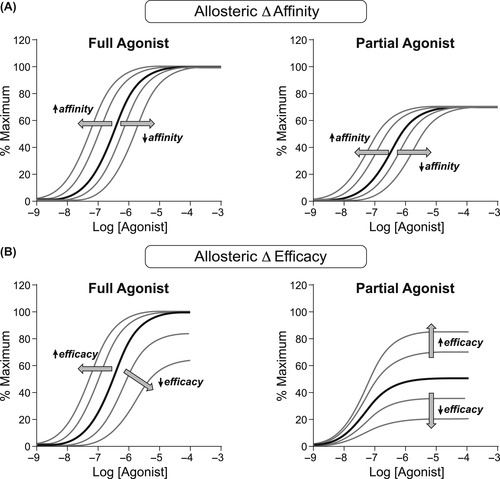
An allosteric effect in a protein basically can change the complete behavior of that protein and the way it interacts with other molecules. Thus, in the case of a receptor and endogenous agonists, an allosteric modulator can change the affinity of the agonist, the efficacy of the agonist, or both, and these changes need not be in the same direction. Figure 5.6A shows the effect of an allosteric modulator on the affinity of full and partial agonists; an increased affinity causes a shift to the left of the dose–response curve, and a decreased affinity causes a shift to the right. Figure 5.6B shows the effect of allosteric changes in the efficacy of full and partial agonists. In this case, an increased efficacy will shift the curve to a full agonist to the left, whereas for a partial agonist it will produce an increase in the maximal response. Similarly, a decreased efficacy can shift the dose–response curve to a full agonist with a high receptor reserve to the right or, in cases of smaller receptor reserves, depress the maximal response. For a partial agonist, which by definition has no receptor reserve, a decrease in efficacy will depress the maximal response (see Fig. 5.6B). At this point it is worth considering how these properties of allosteric modulators can lead to unique drug behaviors therapeutically.