, Arthur H. Cohen2, Robert B. Colvin3, J. Charles Jennette4 and Charles E. Alpers5
(1)
Department of Pathology, Microbiology and Immunology, Vanderbilt University Medical Center, Nashville, Tennessee, USA
(2)
Department of Pathology and Laboratory Medicine, Cedars-Sinai Medical Center, Los Angeles, California, USA
(3)
Department of Pathology Harvard Medical School, Massachusetts General Hospital, Boston, Massachusetts, USA
(4)
Department of Pathology and Laboratory Medicine, University of North Carolina, Chapel Hill, North Carolina, USA
(5)
Department of Pathology, University of Washington, Seattle, Washington, USA
Abstract
The renal transplant biopsy remains the gold standard for the diagnosis of episodes of graft dysfunction that occur in 10–30 % of patients after transplantation [1]. In a prospective trial, the biopsy diagnosis changed the patient management as based on the prebiopsy clinical diagnosis in 42 % of graft dysfunction episodes (39 % in the first month, 56 % in the first year, and 39 % after 1 year); in 19 % unnecessary immunosuppression was avoided [2]. Donor biopsies are used in many centers to get a baseline assessment of the status of the kidney. Protocol biopsies, once used only for research, have become part of the management of the high-risk patient and in some centers for all patients (surveillance biopsies).
The renal transplant biopsy remains the gold standard for the diagnosis of episodes of graft dysfunction that occur in 10–30 % of patients after transplantation [1]. In a prospective trial, the biopsy diagnosis changed the patient management as based on the prebiopsy clinical diagnosis in 42 % of graft dysfunction episodes (39 % in the first month, 56 % in the first year, and 39 % after 1 year); in 19 % unnecessary immunosuppression was avoided [2]. Donor biopsies are used in many centers to get a baseline assessment of the status of the kidney. Protocol biopsies, once used only for research, have become part of the management of the high-risk patient and in some centers for all patients (surveillance biopsies).
The processing of the biopsy is the same as for native kidneys, with the addition of C4d staining for antibody-mediated rejection. In our center we routinely process two cores for histology, and portions of each are frozen and fixed for electron microscopy. Electron microscopy is valuable for the diagnosis of biopsies in which recurrent or de novo glomerular disease is in the differential and in chronic antibody-mediated rejection. An adequate sample includes two cores, because the sensitivity of detecting acute rejection on one core is about 90 %; with two cores the sensitivity is increased to 99 % [3]. We routinely make five levels, three stained with H&E and one each with PAS and trichrome. Immunohistochemistry is used for identification of microorganisms and cell types as indicated. Molecular tests are likely to be applied in the future to refine the diagnosis [1].
Renal allograft biopsies are a challenge for the pathologist, since they may have all the diseases of a native kidney, plus unique diseases seen only in allografts. The urgency for treatment often means a rapid diagnosis is sought, sometimes with only a frozen section, with its known limitations [4].
Furthermore, there are often multiple diseases present, and it is up to the pathologist and clinician to decide which is dominant. On the other hand, transplant biopsies provide a window on the early stages of native kidney diseases and can yield pathogenetic insights. The classification system we use for transplant biopsies is give in Table 20.1 [5].
Table 20.1
Pathologic classification of renal allograft lesions
I. Alloimmune |
A. T-cell-mediated rejection |
1. Acute T-cell-mediated rejection (acute cellular rejection) |
(a) Tubulointerstitial (Banff type I) |
(b) Endarteritis/endothelialitis (Banff type II) |
(c) Arterial transmural inflammation or fibrinoid necrosis (Banff type III) |
(d) Transplant glomerulitis (no Banff type) |
2. Chronic T-cell-mediated rejection |
(a) Tubulointerstitial (Banff type I + IF/TA) |
(b) Transplant arteriopathy (Banff type II) + intimal fibrosis/foam cells |
B. Antibody-mediated rejection |
1. Hyperacute rejection |
2. Acute antibody-mediated rejection (acute humoral rejection) |
(a) Acute tubular injury (Banff type I) |
(b) Capillary inflammation (Banff type II) |
(c) Arterial fibrinoid necrosis (Banff type III) |
3. Chronic antibody-mediated rejection |
(a) Transplant glomerulopathy and glomerulitis |
(b) Peritubular capillary multilamination and capillaritis |
(c) Transplant arteriopathy |
C. Alloreactivity without pathologic evidence of rejection |
1. Accommodation (C4d and or DSA with active rejection) |
2. Treg-rich organized lymphoid structures (TOLS) |
D. Autoantibody-/alloantibody-mediated renal diseases |
1. De novo membranous glomerulonephritis |
2. Anti-angiotensin type II receptor autoantibody syndrome |
E. Recipient-specific alloantibody-mediated diseases |
1. Anti-glomerular basement membrane (GBM) disease (in Alport’s syndrome) |
2. Anti-tubular basement membrane (TBM) disease |
II. Drug toxicity and hypersensitivity (partial list) |
A. Calcineurin inhibitor nephrotoxicity: acute tubulopathy, chronic hyaline arteriolopathy, IFTA, FSGS, thrombotic microangiopathy (TMA) |
B. mTOR inhibitor toxicity (acute tubular injury, FSGS, TMA) |
C. Antiviral drug tubular toxicity |
D. Allergic interstitial nephritis |
III. Infection (viral, bacterial, fungal) |
A. Polyomavirus nephropathy |
B. Adenovirus nephropathy |
C. Posttransplant lymphoproliferative disease |
IV. Ischemia |
A. Acute ischemic injury (ATN) |
B. Perfusion injury |
C. Major artery/vein thrombosis |
D. Renal artery stenosis |
V. Obstruction |
VI. Recurrent primary disease |
A. Immunologic (e.g., IgA nephropathy, lupus nephritis, anti-GBM disease) |
B. Metabolic (e.g., amyloidosis, diabetes, oxalosis) |
C. Unknown etiology (e.g., dense deposit disease, focal segmental glomerulosclerosis) |
VII. De novo glomerular disease (partial list) |
A. Focal segmental glomerulosclerosis (hyperfiltration/collapsing FSGS) |
B. Diabetic nephropathy |
The most widely used classification system is the Banff schema, as modified by the Collaborative Clinical Trials in Transplantation (CCTT) categories [3], and updated biannually [6–9]. The Banff system scores each element (infiltrate, tubulitis, arteritis, glomerulitis) on a 0–3 scale (1, mild; 2, moderate; 3, severe) as the basis for grading. The Banff system promoted standardization of definitions and has created a common language. However, there remain issues to be solved, such as the “borderline” category and glomerular lesions. The interobserver reproducibility for component grading of the Banff system is disappointing, with kappa values of <0.4 (in the poor range) [10]. The scoring for tubulitis and arteritis was improved by scoring photographs, indicating that finding the relevant lesion on the slide was a limiting factor; scoring of the percent infiltrate or glomerulitis was not improved by scoring a photograph, suggesting intrinsic problems in the definitions or methodology.
Acute Cellular Rejection (ACR)
Clinical
Acute cellular rejection (ACR) is the common form of rejection, mediated by T cells, that develops classically 1–6 weeks after transplantation but may erupt at any time. The typical clinical presentation is a rising creatinine over a few days, accompanied by weight gain and sometimes fever and graft tenderness. The frequency of clinical ACR has been reduced with modern therapy to about 10 %.
Pathologic Findings
Light Microscopy
Acute cellular rejection affects the interstitium, tubules, endothelium, and glomeruli, separately or in combination. The characteristic microscopic features of tubulointerstitial rejection (Banff type I) are pleomorphic interstitial infiltrates of activated lymphocytes and monocytes, accompanied by interstitial edema and tubular injury. The cells invade tubules (“tubulitis”) and infiltrate the cortex (10–25 % in the Banff system is considered suspicious for rejection and >25 % is rejection) (Fig. 20.1). The infiltrating mononuclear cells typically include lymphoblasts, with cytoplasmic basophilia, nucleoli, and occasional mitotic figures, indicative of increased synthetic and proliferative activity. The infiltrate consists of T cells (CD4 and CD8), macrophages, and sometimes B cells. Foxp3+ cells (presumptive T regulatory cells) also accumulate in ACR [11] and may be correlated with a better prognosis in protocol biopsies [12]. TGF-β mRNA is associated with better outcome, perhaps related to promotion of Treg cells [13].
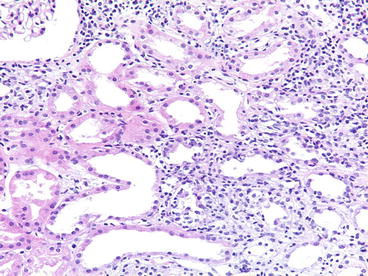
Fig. 20.1
Acute cellular rejection (type I). A diffuse mononuclear infiltrate is present with edema and tubulitis [periodic acid-Schiff (PAS) stain]
Infiltration of mononuclear cells under arterial and arteriolar endothelium is the pathognomic lesion of Banff type II ACR (“endarteritis” or “endothelialitis”) (Fig. 20.2). When lymphocytes are only on the surface of the endothelium, their significance is less certain. Lymphocytes also commonly surround vessels, a nonspecific feature, unless the cells invade the media. Endarteritis has been reported in 20–50 % of the renal biopsies with acute cellular rejection [14]. Some do not find the lesion as often, which may possibly be ascribed to inadequate sampling, overdiagnosis of rejection, or timing of the biopsy with respect to antirejection therapy. These lesions are more frequent in the larger arteries [14]. In our experience, about 20 % of the arteries are involved so that levels of several arteries in the sample are essential for its detection [14]. Rarely, the lymphocytes invade through the media, so-called transmural endarteritis (Fig. 20.3).
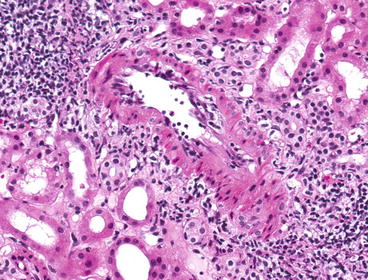
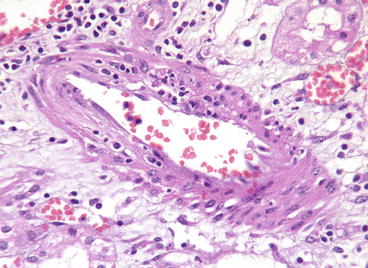
Fig. 20.2
Acute cellular rejection with endarteritis (type II). A small artery has subendothelial infiltration of mononuclear cells (H&E stain)
Fig. 20.3
Acute cellular rejection, type III. Transmural inflammation of an artery is shown. The C4d stain was negative
In most cases of acute cellular rejection, the glomeruli are spared or show minor changes, typically a few scattered mononuclear cells (T cells and monocytes) and minimal segmental endothelial damage, termed “glomerulitis” [15]. Occasionally, a severe, diffuse form of glomerular injury is evident and dominates the histologic pattern (Fig. 20.4). In 1981 Richardson and his colleagues [16] drew attention to a distinctive, acute allograft glomerulopathy, characterized by hypercellularity, injury, and enlargement of endothelial cells, infiltration of glomeruli by mononuclear cells, and webs of periodic acid-Schiff (PAS)-positive material. This severe form of glomerulopathy is observed in ~4 % of biopsies taken for allograft dysfunction, typically 1–4 months after transplantation and is believed to be an unusual variant of cellular rejection, sometimes promoted by cytomegalovirus (CMV) infection [15]. T cells are regularly detected in glomeruli immunohistochemically and OKT3 can reverse the lesion [17]. For unknown reasons, rejection becomes focused on glomerular components; florid glomerulopathy may occur with little interstitial inflammation, although cellular endarteritis is common. Glomerulitis is also commonly seen in antibody-mediated rejection and is a sign of activity. Glomerulitis in AMR is predominately due to macrophages rather than T cells which dominate in ACR [18].
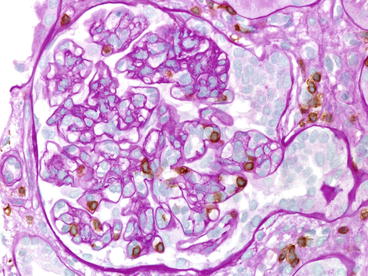
Fig. 20.4
Acute transplant glomerulitis. This pattern of glomerular injury can occur as a feature of cellular or humoral rejection; T cells predominate in cellular rejection, as in this case, which also has a tip lesion (PAS stain with anti-CD3) (Case courtesy of M. Pilichowska (Tufts))
Interstitial mononuclear inflammation and tubulitis occur in a variety of diseases other than acute rejection, such as drug-induced allergic tubulointerstitial nephritis and obstruction, and therefore cannot be considered proof that rejection is present. The presence of systemic infection is another well-known cause of tubulointerstitial nephritis and tubulitis. Tubulitis is often present in atrophic tubules from any cause and does not indicate acute rejection. Posttreatment with anti-T-cell antibodies, the inflammation diminishes in most patients, and this is correlated with successful response to therapy. This type of rejection often responds to steroid pulses and rarely causes graft loss [3, 14, 19, 20].
In all large studies, endarteritis has a worse prognosis than tubulointerstitial rejection [3, 14, 19, 20, 21]. Cases with endarteritis are less responsive to steroid pulses, but do respond to OKT3 or antithymocyte globulin (ATGAM), and have a decreased 1-year survival [14]. Later development of intimal fibrosis is correlated prior type II rejection [19].
Endarteritis must not be confused with necrotizing arteritis, characteristic of humoral rejection (type III). Rarely, transmural cellular inflammation causes frank medial necrosis, but this complication of cellular arteritis can be distinguished from necrotizing arteritis by the heavy mononuclear infiltrate. Regrettably, many still do not distinguish these lesions, regarding all “vascular rejection” as predominately humoral. Endarteritis has a much better rate of reversal than necrotizing arteritis (75 % vs. <25 % 1-year graft survival), which alone justifies their separation [19].
Immunofluorescence Microscopy
Immunofluorescence microscopy shows little, if any, immunoglobulin deposition, but interstitial fibrin is typically present. Fibrin and scant immunoglobulin and C3 deposits may also be found in glomeruli. C4d in peritubular capillaries when present is indicative of a component of antibody-mediated rejection (see below).
Electron Microscopy
Electron microscopy is generally not used for the diagnosis of acute rejection. Studies have shown endothelial injury in peritubular and glomerular capillaries, invasion of lymphocytes into tubules. No immune deposits are detectable. Cases with acute allograft glomerulopathy show markedly reactive endothelial cells, with mononuclear cells in capillaries and often platelet/fibrin aggregates.
Pathogenesis
Type I and II cellular rejections are mediated primarily by T cells and their coconspirators, macrophages, and other cell types, rather than antibodies, since they both occur in the absence of circulating antibody or evidence of complement fixation in the graft. In animals endarteritis occurs in the absence of humoral antibody (e.g., B-cell-deficient mice). In humans, rejection with vascular infiltrates can usually be reversed with antilymphocyte antibodies such as OKT3 or ATG [14, 22]. Both CD8+ and CD4+ cells invade the intima in early grafts, but later CD8+ cells predominate [15, 23]. Arterial endothelium shows increased human leukocyte antigen DR (HLA-DR), intercellular adhesion molecule 1 (ICAM-1), and vascular cell adhesion molecule 1 (VCAM-1) in acute rejection [23], and the tubular epithelium shows increased HLA-DR [24]. Increased expression of major histocompatibility complex (MHC) antigen and adhesion molecules is believed to be a response to cytokines. Microarray and PCR studies have shown a marked increase in gene expression for a variety of molecules of the cytotoxic T cell, macrophages, and molecules induced by interferon gamma [25]. In contrast, mRNA for tubular cell transporter molecules is diminished, a marker of their injury. Practical application of these techniques is expected in the future once their added clinical value is demonstrated [1].
Acute Antibody-Mediated Rejection (Acute AMR) or Acute Humoral Rejection
Clinical
The clinical features of acute AMR consist of severe and acute (days) graft dysfunction that is not responsive to steroids or usually antilymphocyte antibodies and with a higher rate of graft loss than cellular rejection. It can arise anytime after transplant. The risk factors for acute AMR include elevated panel reactive antibody (PRA), prior transplant, and historically positive crossmatch, all associated with prior sensitization [26–29].
Pathologic Findings
Light Microscopy
Three forms of acute AMR have been recognized [6, 30]: (1) acute tubular injury with little inflammation, (2) peritubular and glomerular capillary inflammation with neutrophils, and (3) necrosis of arteries. Acute humoral rejection (AHR) can be missed in routine sections.
In one series, a component of AHR would not have been recognized in 25 % of cases without the C4d stain: 15 % showed only ACR and 10 % only acute tubular injury [30]. The 10 % acute tubular injury biopsies comprised of two donor-specific antibodies (DSA+) in patients with widespread C4d staining of peritubular capillaries (PTCs) that showed predominantly acute tubular injury on initial biopsy. Later biopsies performed in one of these showed typical AHR with abundant neutrophils in PTCs and glomeruli as well as fibrinoid necrosis. Most cases have neutrophils in PTCs (Fig. 20.5). This feature is useful, but not highly sensitive or specific [30]. The histologic features in the Regele et al. series were similarly misleading: 32 % of those with C4d did not meet the Banff criteria for rejection, accounting for about 75 % of the patients with acute AMR [27]. In some cases arterial and arteriolar thrombosis and glomerular thrombosis and necrosis predominate similarly to thrombotic microangiopathy. These lesions must be distinguished from the hemolytic uremic syndrome of calcineurin inhibitor toxicity, which can also have thrombi in arterioles and glomeruli, but does not affect the peritubular capillaries (or have C4d). A minority of cases have fibrinoid necrosis, in which the arterial media shows myocyte necrosis, fragmentation of elastica, and accumulation of brightly eosinophilic material called “fibrinoid” and little mononuclear infiltrate in the intima or adventitia (Fig. 20.6). A scant infiltrate of neutrophils and eosinophils and thrombosis may be present.
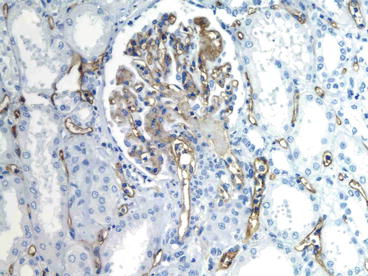
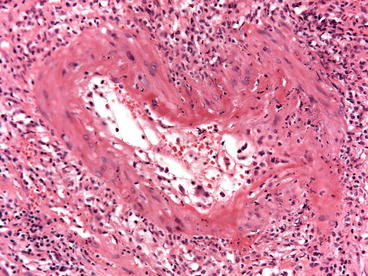
Fig. 20.5
Acute antibody-mediated rejection, type II. Neutrophils are in the peritubular capillaries (capillaritis) and in the glomeruli. The peritubular and glomerular capillaries stain strongly for C4d (immunohistochemistry, anti-C4d)
Fig. 20.6
Acute AMR with fibrinoid necrosis (type III). A small artery has fibrin, neutrophils, and necrosis in the media (H&E stain)
Immunofluorescence Microscopy
Detection of C4d has proved to be useful in the detection of humoral rejection (Fig. 20.7). After antibody binds to antigen, C4 is proteolytically cleaved by activated C1 into C4a and C4b. The cleavage of C4 exposes the reactive and short-lived thiol group in C4b that binds covalently to nearby molecules containing amino or hydroxyl groups, such as proteins and carbohydrates [31]. Bound C4b is proteolytically inactivated into C4d, a 44.5-kd peptide, which contains the thioester site and remains covalently bound at the same site. Thus, if C4d is bound to a structural protein, it is potentially a durable marker of local complement activation by the classical pathway. C4d can disappear as soon as 8 days [32] and after 60 days is usually negative (unless chronic humoral rejection develops) [30]. Cases with fibrinoid necrosis of arteries typically contain immunoglobulin (Ig, usually IgG and IgM), C3, and fibrin in the arterial walls. The criteria for acute humoral rejection proposed by Mauiyyedi et al. [30] and accepted with minor modification by the Banff conference are given in Table 20.2 [6].
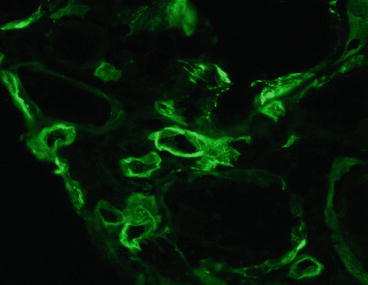
Fig. 20.7
Acute humoral rejection. C4d is present in peritubular capillaries (immunofluorescence on frozen section; monoclonal antibody to C4d)
Table 20.2
Criteria for acute antibody-mediated rejection
1. Widespread, linear C4d+ staining of peritubular capillaries |
2. Evidence for acute renal injury consisting of at least one of the following: |
(a) Acute tubular injury |
(b) Neutrophils in peritubular or glomerular capillaries |
(c) Fibrinoid necrosis of arteries |
3. Circulating anti-donor-specific antibodies |
Pathogenesis
Donor-specific anti-HLA antibodies are detected in 90 % of C4d+ acute rejection cases compared with 2 % in C4d− acute rejection cases (p < 0.001) [30]. We have speculated that the DSA-negative cases are due to adsorption in the graft or to non-HLA specificities. Circulating pretransplant and posttransplant DSA can be identified by T- and B-cell cytotoxicity assays or flow cytometry but best by solid-phase assay with single antigen beads.
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


