Fig. 13.1
Superoxide production in the mitochondrial electron transport chain. The production of superoxide (O2 •−) is thought to occur at Complex I (NADH dehydrogenase), Complex II (succinate dehydrogenase), or Complex III (coenzyme Q-cytochrome c reductase), but under normal metabolic conditions, Complex III is thought to be the main sites of superoxide production. Superoxide from Complex I and Complex II is released into the matrix, whereas superoxide from Complex III is released into the intermembrane space or matrix. OMM=outer mitochondrial membrane, IMM=inner mitochondrial membrane)
Mitochondria are protected against ROS by an interacting network of antioxidant enzymes (Table 13.1) (Balaban et al. 2005; Halliwell and Gutteridge 2007; Finkel and Holbrook 2000; West et al. 2011). SOD2 converts superoxide into hydrogen peroxide, which in turn is decomposed to water by two major mitochondrial antioxidant defense systems: the glutathione and thioredoxin systems (Halliwell and Gutteridge 2007; Mari et al. 2009; Ribas et al. 2014; Aon et al. 2012). There are three major players in the mitochondrial glutathione system: glutathione, glutathione peroxidase 1 (GPX1), and glutathione reductase (GSR). Glutathione acts as the major antioxidant in cells. When glutathione is in a reduced state or an active form, it interacts with GPX1 to decompose hydrogen peroxide into water. NADPH-dependent GSR then reduces oxidized glutathione (GSSG) to reduced glutathione (GSH). There are three major players in the mitochondrial thioredoxin system: thioredoxin 2 (TXN2), peroxiredoxin 3 (PRDX3), and thioredoxin reductase 2 (TXNRD2). When TXN2 is in a reduced state, it interacts with PRDX3 to decompose hydrogen peroxide into water. NADPH-dependent TXNRD2 then regenerates reduced TXN2 (TXN2red) from oxidized TXN2 (TXN2oxi). These antioxidant defense systems work with each other to protect key mitochondrial components such as mtDNA, proteins, and membranes from ROS-induced oxidative damage (Evans and Halliwell 1999). It is thought that the mitochondrial antioxidant defense systems do not keep pace with the age-related increase in mitochondrial ROS production, and that during aging, the balance between the mitochondrial antioxidant defense and ROS production shifts progressively toward a more pro-oxidant state (Rebrin and Sohal 2008). Therefore, the balance between ROS production and mitochondrial antioxidant defense capacity may determine the degree of ROS-induced oxidative damage and associated mitochondrial dysfunction in the inner ear sensory hair cells or spiral ganglion neurons, which require larger amounts of energy to process sounds. In this chapter, we summarize what has been learned about the mitochondrial biochemical pathways and/or targets of age-related hearing loss (AHL), and examine the link between aging, mitochondrial dysfunction, and hearing loss in rodents and humans.
Table 13.1
Mitochondrial antioxidant defense enzymes
Enzyme | Enzymatic reaction |
|---|---|
SOD2 | 2O2 •− + 2H+ → H2O2 + O2 |
GPX1 | H2O2 + 2GSH → GSSG + 2H2O |
GSR | GSSG + NADPH + H+ → 2GSH + NADP+ |
PRDX3 | H2O2 + TXN2red → TXN2oxi + 2H2O |
TXNRD2 | TXN2oxi + NADPH + H+ → TXN2red + NADP+ |
13.2 Molecular Targets of AHL Involved in the Maintenance of Mitochondrial Function
13.2.1 Citrate Synthase
Citrate synthase (CS) is the first and rate-limiting enzyme of the tricarboxylic acid (TCA) cycle in the mitochondria (Johnson et al. 2012: Raimundo et al. 2011). CS is encoded by a single nuclear gene and is localized in the mitochondrial matrix where it functions as the first step of the TCA cycle, producing citric acid from acetyl-CoA and oxaloacetate (Fig. 13.2). Hence, CS plays a critical role in energy metabolism. Johnson et al. mapped an ahl4 locus on Chromosome 10 that contributes to AHL of the A/J mouse strain (Johnson et al. 2012), and found that a CS mutation underlies ahl4-related hearing loss in the A/J strain. These A/J mice display early onset and rapid progression of hearing loss. The authors sequenced the entire genomes of A/J and four strains that lack an ahl4 phenotype, and found that the rs29358506 single nucleotide polymorphism (SNP), located in exon 3 of CS, is the only exon or splice site variant within the 5.5 megabases (Mb) candidate region that is unique to the A/J strain, indicating that CS is the gene responsible for ahl4. Consistent with these results, A/J mice display poor performance in endurance exercise such as having the shortest treadmill duration time among ten inbred mouse strains, including the C57BL/6 strain (Lightfoot et al. 2001). Another study has also shown poor treadmill performance in A/J mice, estimating the endurance capacity of BALB/c mice to be 217 % greater than that of A/J mice (Haramizu et al. 2009). Collectively, these results support the importance of CS in energy metabolism and are consistent with the suggestion that a decline in CS activity and resulting decreased ATP production in the inner ear tissues may lead to mitochondria dysfunction and associated cochlear cell death.
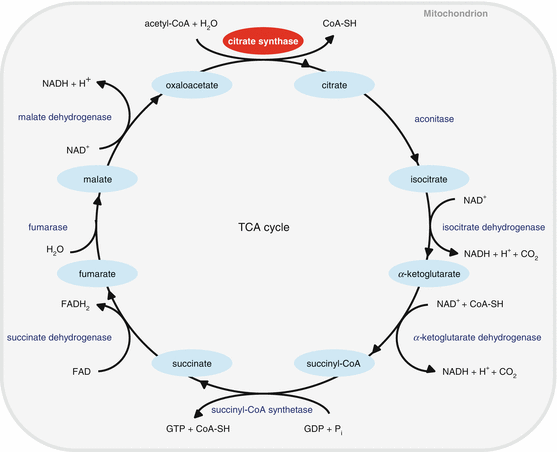
Fig. 13.2
TCA cycle. Citrate synthase (CS) is the first and rate-limiting enzyme of the TCA cycle in the mitochondria
13.2.2 Uncoupling Protein 2
Uncoupling protein 2 (UCP2) is a member of the mitochondrial anion carrier protein family, which facilitates the transfer of anions from the inner to the outer mitochondrial membrane and the return transfer of protons from the outer to the inner mitochondrial membrane (Fig. 13.3) (Sugiura et al. 2010; Mattiasson et al. 2003). UCPs also reduce the mitochondrial membrane potential in mammalian cells. UCP2 is expressed in multiple tissues, including skeletal muscles and central nervous system and is thought to play a role in thermogenesis, obesity, and diabetes. A neuroprotective role for UCP2 was indicated by the finding that brain damage following experimental stroke was reduced in mice overexpressing human UCP2 (Mattiasson et al. 2003). Moreover, UCP2, UCP3, and UCP4 mRNAs were found to be expressed in the vestibular and spiral ganglions in the inner ear, while UCP3 mRNA expression was undetectable in the brain of rats (Kitahara et al. 2004). A human epidemiological study in a Japanese population discovered an association between the UCP1 A-3826G polymorphism (rs1800592) and obesity (Sugiura et al. 2010). Furthermore, Sugiura et al. investigated the association between UCP1 and UCP2 gene polymorphisms and hearing impairment in middle-aged and elderly males and females in Japan (Sugiura et al. 2010). Detailed questionnaires, pure-tone audiometry measurements, and UCP1 A-3826G and UCP2 Ala55Val polymorphisms were examined in 1,547 subjects between the ages of 40 and 79 years. Using generalized estimating equations, associations between hearing impairment and the gene polymorphisms in UCP1 and UCP2 with age, sex, history of occupational noise exposure, and body mass index were analyzed under dominant, recessive, and additive models. Sugiura et al. found that UCP2 Ala55Val polymorphisms exhibited a significant association with AHL in the Japanese population. Together, these reports suggest that a decline in mitochondrial UCP2 activity may accelerate the progression of AHL in humans.
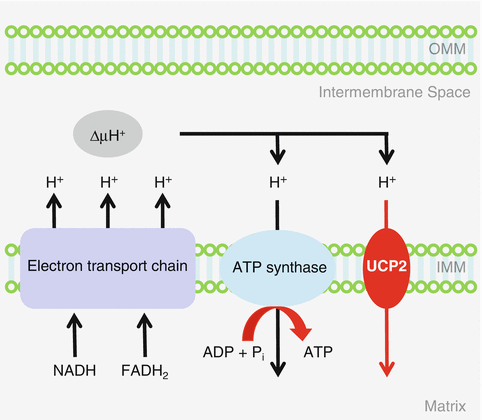
Fig. 13.3
Mitochondrial uncoupling. Uncoupling protein 2 (UCP2) is a member of the mitochondrial anion carrier protein family, which facilitates the transfer of anions from the inner to the outer mitochondrial membrane and the return transfer of protons from the outer to the inner mitochondrial membrane. OMM=outer mitochondrial membrane, IMM=inner mitochondrial membrane
13.2.3 Mitochondrial DNA Polymerase Gamma
mtDNA POLG is the only DNA polymerase that is active in the mitochondria and that can proofread and replicate mtDNA (Fig. 13.4) (Kujoth et al. 2007). Polg knockin mice were created by introducing a two-base substitution, which results in a defect in mtDNA proofreading ability (Kujoth et al. 2005; Trifunovic et al. 2004). Young Polg mutator mice were indistinguishable from wild-type (WT) littermates, but these mutator mice displayed a variety of premature aging phenotypes such as kyphosis and alopecia by 9 months of age. More importantly, Polg mutator mice displayed a reduced life span: the median survival of the Polg mutator mice was 416 days, while the median life span of WT mice was >850 days. Age-related weight loss, a common feature of the elderly, was noted from 24 weeks of age in these animals. Nine months old mutator mice also displayed reduced bone mineral density, a common feature of age-related osteoporosis and age-related loss of skeletal muscle, a common feature of sarcopenia. Auditory brainstem response (ABR) hearing tests were conducted in these animals: young Polg mutator mice displayed normal hearing compared to age-matched WT mice: however, by 9–10 months of age, Polg mutator mice displayed a significant elevation of ABR thresholds at 4, 8, and 16 kHz (Kujoth et al. 2005; Someya et al. 2008; Niu et al. 2007). Examination of the histology of the basal cochlear region confirmed that 9–10-month-old Polg mutator mice displayed severe loss of spiral ganglion neurons and hair cells (Kujoth et al. 2007; Someya et al. 2008; Niu et al. 2007). These animal study results were consistent with the observation that a defect in the human POLG gene causes Alper’s syndrome that is associated with both mitochondrial dysfunction and hearing loss (Kujoth et al. 2007; Someya and Prolla 2010; Mancuso et al. 2004). Together, these reports suggest that a decline in POLG activity in the mitochondria causes mitochondrial dysfunction and associated cochlear cell loss, leading to the progression of AHL.
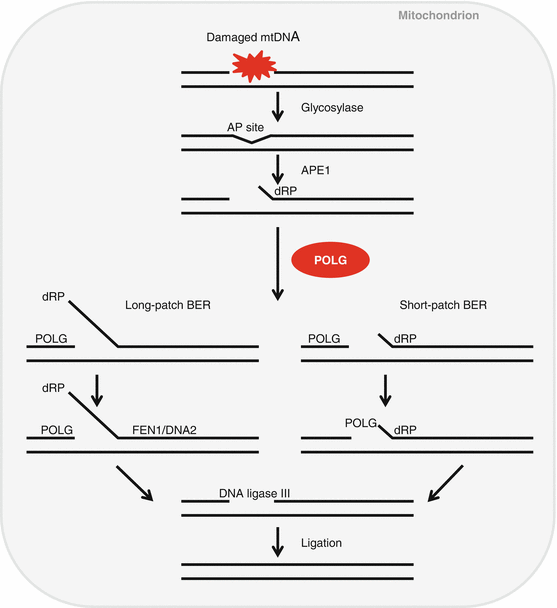
Fig. 13.4
Mitochondrial DNA repair. Mitochondrial DNA polymerase gamma (POLG) is the only DNA polymerase that is active in the mitochondria and that can proofread/repair and replicate mtDNA. AP site (apurinic/apyrimidinic (AP) site), APE1 (AP endonuclease 1), dRP (5’ 2-deoxyribose 5-phosphate), BER (base excision repair), FEN1 (flap endonuclease 1), DNA2 (DNA replication ATP-dependent helicase/nuclease)
13.2.4 Glutathione Peroxidase 1
Glutathione peroxidase 1 (GPX1) plays an important role in mitochondrial antioxidant defense by decomposing hydrogen peroxide into water (Fig. 13.5) (Halliwell and Gutteridge 2007; Mari et al. 2009). A previous study has shown that cochlear ROS levels were elevated following acute noise exposure in C57BL/6 mice (Ohlemiller et al. 1999). Consistent with this report, mice lacking superoxide dismutase 1 (Sod1) displayed increased hair cell loss in the cochlea compared to WT mice (McFadden et al. 1999). Oxidative protein damage also increases with age in the cochleae of CBA/J mice (Jiang et al. 2007), while oxidative nuclear DNA damage increases with age in the cochlea of C57BL/6 mice (Someya et al. 2009). Ohlemiller et al. (2000) investigated whether Gpx1 deficiency increases susceptibility to noise-induced hearing loss (NIHL) in mice and found that Gpx1 −/− mice showed significantly greater ABR threshold elevation after noise exposure compared to WT mice. Noise-exposed Gpx1 −/− mice also showed more sensory hair cell loss compared to WT mice. These results suggest that a decline in GPX1 activity in the mitochondria may lead to increased levels of ROS, which in turn results in mitochondrial dysfunction, leading to cochlear cell loss and associated hearing loss.
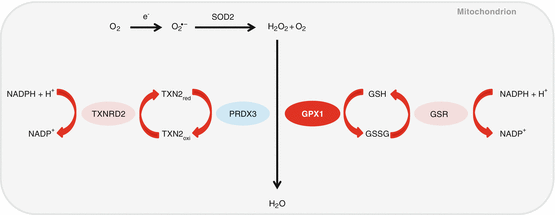
Fig. 13.5
Mitochondrial antioxidant defense system. Glutathione peroxidase 1 (GPX1) plays an important role in mitochondrial antioxidant defense by decomposing hydrogen peroxide into water. TXNRD2=thioredoxin reductase 2, PRDX3=peroxiredoxin 3, GPX1=glutathione peroxidase 1
13.2.5 Catalase
Catalase (CAT) converts hydrogen peroxide into water and oxygen, and is one of the most efficient enzymes found in cells (Fig. 13.6) (Halliwell and Gutteridge 2007): Each catalase molecule can decompose millions of hydrogen peroxide molecules every second, indicating the importance of this antioxidant enzyme in protecting the cells against ROS. Catalase is usually localized in the peroxisomes, although catalase expression was also observed in the cytosol and mitochondria. Schriner et al. generated mice overexpressing human catalase targeted to the peroxisome (PCAT), nucleus (NCAT), or mitochondria (MCAT) and found that overexpression of catalase in the mitochondria extended both median and maximum life span in both males and females, while overexpression of catalase in the nuclei did not show a significant extension of median nor maximum life span (Schriner et al. 2005). PCAT animals showed a slight extension of median life span. MCAT mice also displayed increased CAT activities in the heart, skeletal muscle, and brain, reduced oxidative DNA damage, and decreased hydrogen peroxide levels in the heart. In agreement with these reports, young MCAT mice display normal hearing compared to age-matched WT mice: however, middle-aged MCAT mice display significantly lower ABR thresholds at 8, 16, and 32 kHz than those of middle-aged WT mice. Furthermore, MCAT mice display reduced oxidative DNA damage and loss of spiral ganglion neurons and hair cells in the cochlea (Someya et al. 2009). Importantly, in humans, CAT activity was significantly higher in the red blood cells of centenarians compared to young healthy adults (Kłapcińska et al. 2000). Collectively, these results suggest that enhancing CAT activity and/or reducing hydrogen peroxide levels in cochlear mitochondria may slow the development of age-related cochlear degeneration and hearing loss.
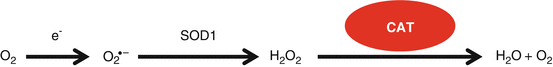
Fig. 13.6
Peroxisomal/cytosolic antioxidant defense system. Catalase (CAT) is localized in the peroxisome or cytosol and converts hydrogen peroxide into water and oxygen in the cells
13.2.6 BCL-2-Antagonist/Killer
It is well-documented that mitochondria play a major role in apoptosis which is regulated by BCL-2 (b-cell lymphoma 2) family members (Mattson 2000; Youle and Strasser 2008; Lindsten et al. 2000). Of the BCL-2 family members, the proapoptotic proteins BCL-2 antagonist/killer (BAK) and BCL-2-associated X (BAX) play a central role in promoting mitochondrial-mediated apoptosis. These proapoptotic proteins promote permeabilization of the outer mitochondrial membrane, leading to caspase activation and cell death (Fig. 13.7). Lindsten et al. generated Bak-deficient mice and found that homozygous Bak −/− mice are fertile and do not display any developmental abnormalities, suggesting that the role of BAK in development may be redundant with that of other proapoptotic BCL-2 family members such as BAX (Lindsten et al. 2000). We investigated whether BAX and BAK may function in a redundant manner in the auditory system and found no significant differences between middle-aged WT and Bax −/− mice; both had similar hearing loss indicated by higher ABR thresholds and the lower numbers of hair cells and spiral ganglion neurons typically seen in middle-aged C57BL/6 mice (Someya et al. 2009). In contrast, middle-aged Bak −/− mice displayed significantly lower ABR thresholds at 8, 16, and 32 kHz and fewer losses of spiral ganglion neurons and hair cells than those of age-matched WT. Moreover, TUNEL (terminal deoxynucleotidyl transferase dUTP nick end labeling) staining revealed that the levels of apoptotic nuclear fragmentation markers were significantly reduced in the cochlea of middle-aged Bak −/− mice compared to age-matched WT mice. Therefore, a decline in BAK-mediated mitochondrial apoptosis may prevent mitochondrial dysfunction, which in turn prevents cochlear cell death, and the progression of hearing loss during aging.
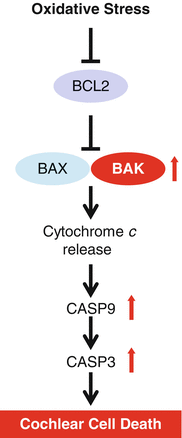
Fig. 13.7
Mitochondrial apoptosis. BCL-2-antagonist/killer (BAK) promotes permeabilization of the outer mitochondrial membrane, leading to caspase activation and cell death. CASP9=caspase-9, CASP3=caspase-3
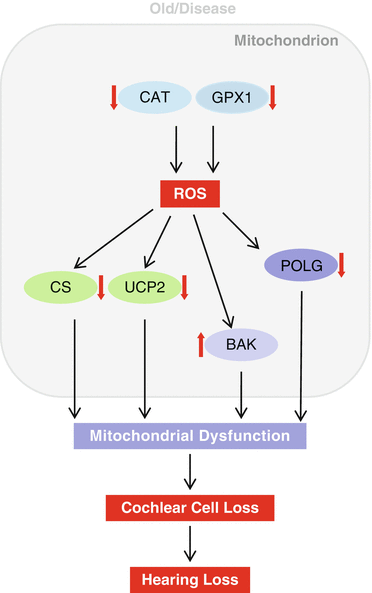
13.3 Conclusions
We have reviewed what has been learned about the mitochondrial biochemical pathways associated with AHL, including the TCA cycle, mitochondrial uncoupling, mtDNA repair, mitochondrial antioxidant defense, and mitochondrial apoptosis pathways (Fig. 13.8), and examined the link between aging, mitochondrial dysfunction, and hearing loss in rodents and humans. Accumulating evidence indicates that mitochondrial dysfunction plays a key role in the development of age-related neurodegenerative diseases, including Alzheimer’s disease (AD), Parkinson’s disease (PD), and amyotrophic lateral sclerosis (ALS) (Finkel and Holbrook 2000; Mari et al. 2009; Mattiasson et al. 2003). AD is characterized by progressive impairment of cognition and emotional disturbances that are associated with synaptic degeneration and loss of neurons (Mattiasson et al. 2003). Increased oxidative damage has been found in the brain of AD patients (Haass and Selkoe 2007), while loss of total glutathione as well as altered glutathione redox status has been observed in the brain of AD individuals (Mari et al. 2009; Boyd-Kimball et al. 2005). PD is characterized by profound motor dysfunction due to degeneration of dopamine neurons in the substantia nigra of the brain (Mattiasson et al. 2003). The early biochemical features of PD include increased mitochondrial dysfunction, oxidative damage, and loss of total glutathione in the dopamine neurons (Perry and Yong 1986). ALS is characterized by progressive paralysis due to degeneration of motor neurons in the spinal cord, and mutations in the SOD1 gene are responsible for some inherited cases of ALS (Mattiasson et al. 2003; Gurney et al. 1994; Wong et al. 1995). Alper’s syndrome, caused by mutations in POLG, is a progressive degenerative disease of the central nervous system that is associated with progressive sclerosing poliodystrophy and deafness (Someya and Prolla 2010; Kujoth et al. 2007; Mancuso et al. 2004). Individuals with Alper’s syndrome have fewer copies of mtDNA in the tissues affected by the disease. This mtDNA depletion leads to impaired oxidative phosphorylation and a decrease in cellular energy, which in turn affect postmitotic tissues such as brain, muscle, and inner ear tissue. It is thought that brain, muscle, and inner ear tissues are most affected because those tissues require larger amounts of energy, are more dependent on oxidative phosphorylation for energy, and damaged cells in these non-regenerative tissues are not generally replaced by new cells, resulting in the variety of symptoms of Alper’s syndrome. Therefore, we speculate that a decline in the activities of mitochondrial enzymes or proteins such as POLG, CS, or UCP2 could play a key role in mitochondrial dysfunction, which in turn leads to development of human age-related progressive hearing loss (Fig. 13.8).