7
Adrenergic, Dopaminergic, and Serotonergic Pharmacology
This chapter is a combination of Chapter 12 Adrenergic Agonists and Antagonists and Chapter 13 5-Hydroxytryptamine (Serotonin) from Goodman and Gilman’s The Pharmacological Basis of Therapeutics, 12th Edition. An understanding of the material in those chapters will be helpful in following the material presented in this chapter. Further details of the therapeutic use of the drugs mentioned in this chapter can be found in subsequent chapters. In addition to the material presented here the above chapters in the 12th Edition include:
• Table 12-1 Chemical Structures and Main Clinical Uses of Important Sympathomimetic Drugs
• A detailed discussion of the pharmacology of the endogenous catecholamines epinephrine (E), norepinephrine (NE), and dopamine (DA)
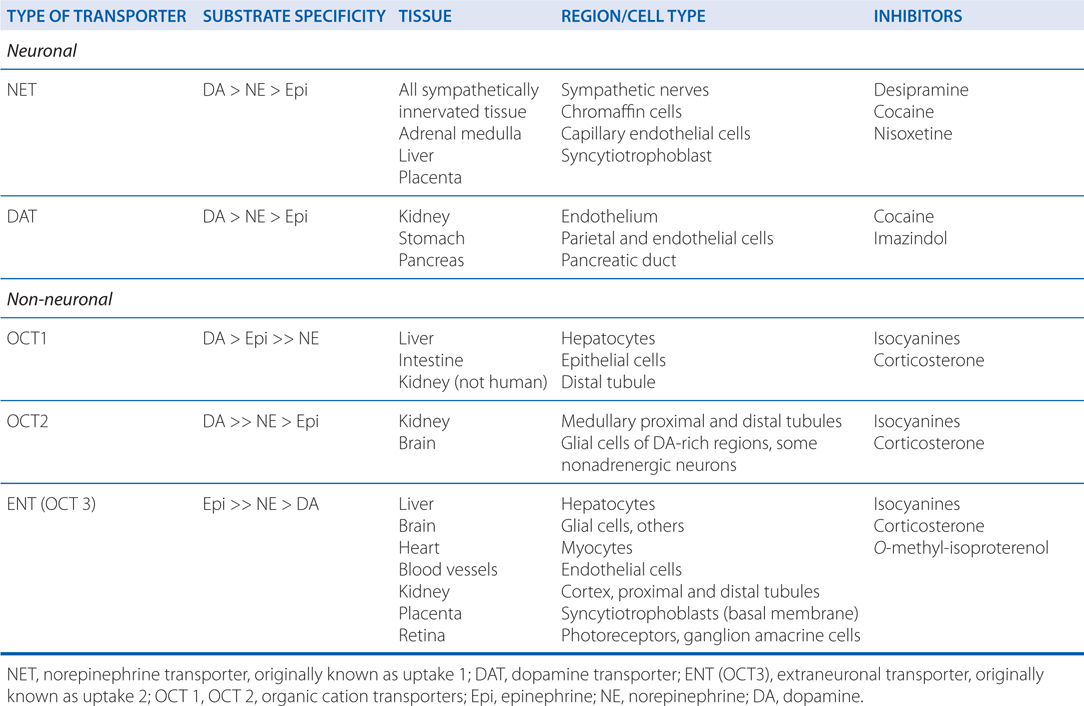
• The pharmacology of miscellaneous sympathomimetic amines, including amphetamine, methamphetamine, methylphenidate, and ephedrine
• A discussion of the therapeutic uses of sympathomimetic drugs in shock, hypotension, cardiac arrhythmias, nasal decongestion, allergic reactions, narcolepsy, weight reduction, and attention deficit/hyperactivity disorder (ADHD)
• Details of the physiological functions of serotonin, including the multiple receptor subtypes of 5-HT
• A discussion of the pharmacology of ergot and the ergot alkaloids, including lysergic acid diethylamide (LSD)
• The physiological functions of dopamine, including a detailed discussion of dopamine receptor subtypes
LEARNING OBJECTIVES
 Understand the pharmacology of the major adrenergic and serotonergic agonists and antagonists.
Understand the pharmacology of the major adrenergic and serotonergic agonists and antagonists.
 Know the mechanisms of action of the adrenergic and serotonergic agonists and antagonists.
Know the mechanisms of action of the adrenergic and serotonergic agonists and antagonists.
 Know the therapeutic uses, adverse effects, toxicities, and contraindications of the adrenergic and serotonergic agonists and antagonists.
Know the therapeutic uses, adverse effects, toxicities, and contraindications of the adrenergic and serotonergic agonists and antagonists.
DRUGS INCLUDED IN THIS CHAPTER
• Acebutolol (SECTRAL, others)
• Albuterol (VENTOLIN-HFA, PROVENTIL-HFA, others)
• Alfuzosin (UROXATRAL)
• Almotriptan (AXERT)
• Alosetron (LOTRONEX)
• Apomorphine (APOKYN)
• Apraclonidine (IOPIDINE)
• Arformeterol (BROVENA)
• Aripiprazole (ABILIFY)
• Atenolol (TENORMIN, others)
• Betaxolog (BETAOPTIC, others)
• Betaxolol (LOKREN, KERCONE, others)
• Bitolterol (TORNALATE)—discontinued in the United States
• Brimonidine (ALPHAGAN, others)
• Bromocriptine (PARLODEL)
• Bucindolol (SANDORMIN)
• Bunazosin—not available in the United States
• Buspirone (BUSPAR, others)
• Cabergoline (DOSTINEX)
• Carteolol (OCCUPRES, others)
• Carvedilol (COREG)
• Celiprolol (SELECTOL)
• Citralopam (CELEXA)
• Clonidine (CATAPRES, others; Transdermal, CATAPRES-TTS)
• Clozapine (FAZACLO)
• Desvenlafaxine (PRISTIQ)
• Dexmedetomide (PRECEDEX)
• Dexmethylphenidate (FOCALIN, FOCALIN XR)
• Dextroamphetamine (DEXIDRINE, ADDERAL XR, others)
• Dobutamine (DOBUTREX)
• Dolasetron (ANZEMET)
• Dopamine
• Dopexamine (DOPACARD)—not currently available in the United States
• Doxazosin (CARDURA, others)
• Duloxetine (CYMBALTA)
• Eletriptan (RELPAX)
• Ephedrine
• Epinephrine (solution for injection, EPIPEN)
• Escitralopam (LEXAPRO)
• Esmolol (BREVIBLOC, others)
• Fenoldopam (CORLOPAM)
• Fenoterol (BEROTEC)—withdrawn from market
• Fluoxetine (PROZAC, others)
• Fluvoxamine (LUVOX, others)
• Formoterol (FORADIL, others)
• Frovatriptan (FROVAL)
• Granisetron (KYTRIL, others)
• Guanabenx (WYTENSIN)
• Guanfacine (TENEX, others; INTUNIV, sustained release)
• Indoramin—not available in the United States
• Isocarboxazid (MARPLAN)
• Isoetharine (BRONKOSOL)—no longer marketed in the United States
• Isoproterenol (ISUPREL)
• Ketanserin (SUFREXAL)—not available in the United States
• Levalbuterol (XOPENEX)
• Levobetaxolol (BETAXON, others)
• Levobunolol (BETAGAN, others)
• Lisdexamfetamine (VYVANASE)
• Mephentermine—discontinued in the United States
• Metaproterenol (ALUPENT, orciprenaline in Europe)
• Metaraminol
• Methamphetamine (DESOXYN)
• Methyldopa (ALDOMET, others)
• Methylphenidate (RITALIN, RITALIN SR, CONCERTA, META-DATE, others; DAYTRANA, transdermal)
• Methylsergide (SANSERT)
• Metipranolol (OPTIPRANOLOL, others)
• Metoprolol (LOPRESSOR, others)
• Midodrine (PROAMATINE, others)
• Milnacipran (SAVELLA)
• Modafinil (PROVIGIL)
• Naphazoline (PRIVINE, NAPHCON, others)
• Naratriptan (AMERGE)
• Nebivolol (NEBILET, BYSTOLIC)
• Norepinephrine (LEVOPHED)
• Olanzapine (ZYPREXA)
• Ondansetron (ZOFRAN, others)
• Oxymetazoline (AFRIN, OCU-CLEAR, others)
• Palonosetron (ALOXI)
• Paroxetine (PAXIL)
• Pemoline (CYLERT, others)—discontinued in the United States
• Pergolide (PERMAX)
• Phenelzine (NARDIL)
• Phenoxybenzamine (DIBENZYLINE)
• Phentolamine (ORAVERSE)
• Phenylephrine (NEO-SYNEPHRINE, others)
• Pindolol (VISKIN, others)
• Pirbuterol (MAXAIR)
• Pramipexole (MIRAPEX)
• Prazosin (MINIPRES, others)
• Procaterol (MASCACIN)—not available in the United States
• Propranolol (INDERAL, INDERAL LA, others)
• Propylhexedrine (BENZEDREX, others)
• Pseudoephedrine (SUDAPHED, others)
• Quetiapine (SEROQUEL)
• Respiridone (RISPERDAL, others)
• Ritodrine—not available in the United States
• Rizatriptan (MAXALT, others)
• Ropinirole (REQUIP)
• Rotigotine (NEUPRO)
• Salmeterol (SEREVENT)
• Sertraline (ZOLOFT)
• Sibutramine (MERIDIA)
• Silodosin (RAPAFLO)
• Sumatriptan (IMITREX, others)
• Tamsulosin (FLOMAX)
• Terazosin (HYTRIN, others)
• Terbutaline (BRETHINE)
• Timolol (TIMOPTIC, others, for ophthalmic use; BLOCADREN, others, for systemic use)
• Tizanidine (ZANAFLEX, others)
• Tranylcypromine (PARNATE, others)
• Urapidil—not available in the United States
• Venlafaxine (EFFEXOR)
• Xylometazoline (OTRIVIN, others)
• Yohimbine (YOCON, APHRODYNE)
• Zolmitriptan (ZOMIG)
MECHANISM OF ACTION OF ENDOGENOUS CATECHOLAMINES
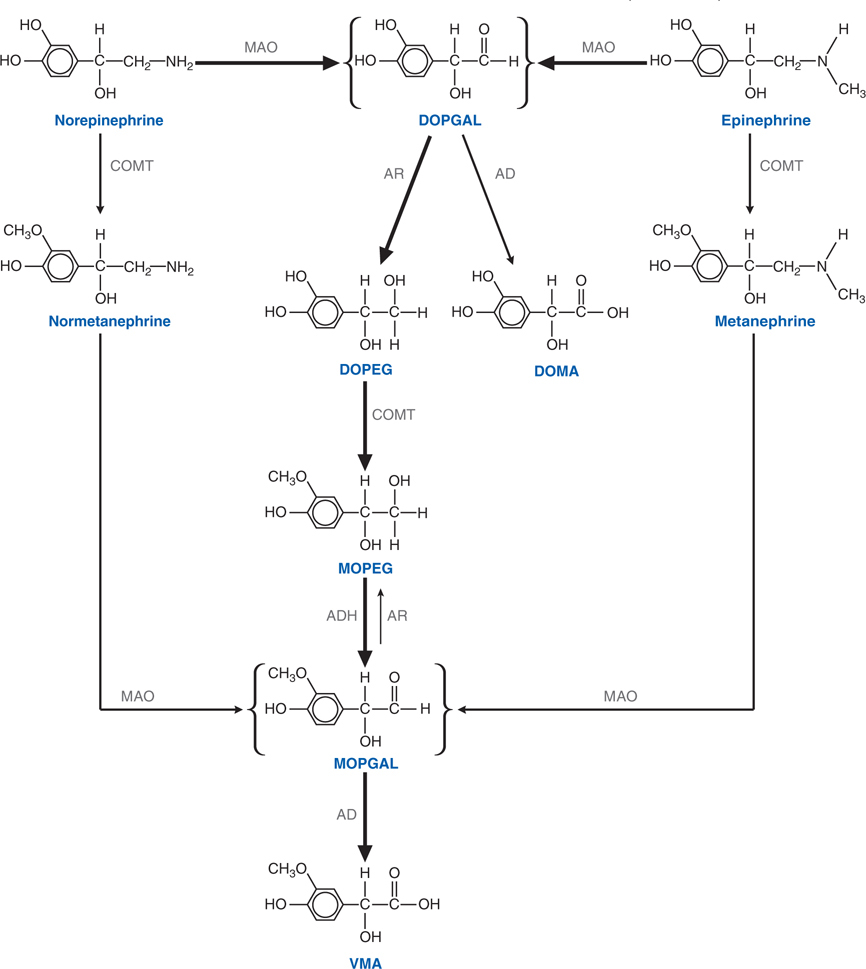
FIGURE 7-1 Steps in the metabolic disposition of catecholamines. Norepinephrine and epinephrine are first oxidatively deaminated to a short-lived intermediate (DOPGAL) by monoamine oxidase (MAO). DOPGAL then undergoes further metabolism to more stable alcohol or acid deaminated metabolites. Aldehyde dehydrogenase (AD) metabolizes DOPGAL to 3,4-dihydroxymandelic acid (DOMA) while aldehyde reductase (AR) metabolizes DOPGAL to 3,4-dihydroxyphenyl glycol (DOPEG). Under normal circumstances DOMA is a minor metabolite with DOPEG being the major metabolite produced from norepinephrine and epinephrine. Once DOPEG leaves the major sites of its formation (sympathetic nerves; adrenal medulla), it is converted to 3-methoxy, 4-hydroxyphenylglycol (MOPEG) by catechol-0-methyl transferase (COMT). MOPEG is then converted to the unstable aldehyde (MOPGAL) by alcohol dehydrogenase (ADH) and finally to vanillyl mandelic acid (VMA) by aldehyde dehydrogenase. VMA is the major end product of norepinephrine and epinephrine metabolism. Another route for the formation of VMA is conversion of norepinephrine or epinephrine into normetanephrine or metanephrine by COMT either in the adreneal medulla or extraneuronal sites, with subsequent metabolism to MOPGAL and thence to VMA.
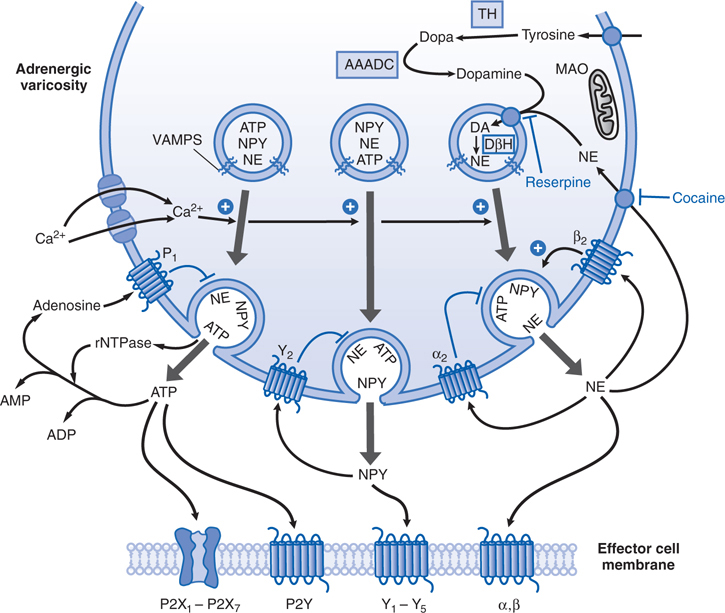
FIGURE 7-2 An adrenergic neuroeffector junction showing features of the synthesis, storage, release, and receptors for norepinephrine (NE), the cotransmitters neuropeptide Y (NPY), and ATP. Tyrosine is transported into the varicosity and is converted to DOPA by tyrosine hydroxylase (TH) and DOPA to dopamine (DA) by the action of aromatic L-amino acid decarboxylase (AAADC). Dopamine is taken up into the vesicles of the varicosity by a transporter, VMAT2, that can be blocked by reserpine. Cytoplasmic NE also can be taken up by this transporter. Dopamine is converted to NE within the vesicle via the action of dopamine-β-hydroxylase (DβH). NE is stored in vesicles along with other cotransmitters, NPY and ATP, depending on the particular neuroeffector junction. Release of the transmitters occurs upon depolarization of the varicosity, which allows entry of Ca2+ through voltage-dependent Ca2+ channels. Elevated levels of Ca2+ promote the fusion of the vesicular membrane with the membrane of the varicosity, with subsequent exocytosis of transmitters. This fusion process involves the interaction of specialized proteins associated with the vesicular membrane (VAMPs, vesicle-associated membrane proteins) and the membrane of the varicosity (SNAPs, synaptosome-associated proteins). In this schematic representation, NE, NPY, and ATP are stored in the same vesicles. Different populations of vesicles, however, may preferentially store different proportions of the cotransmitters. Once in the synapse, NE can interact with α and β adrenergic receptors to produce the characteristic response of the effector. The adrenergic receptors are GPCRs. α and β Receptors also can be located presynaptically where NE can either diminish (α2), or facilitate (β) its own release and that of the cotransmitters. The principal mechanism by which NE is cleared from the synapse is via a cocaine-sensitive neuronal uptake transporter, NET. Once transported into the cytosol, NE can be restored in the vesicle or metabolized by monoamine oxidase (MAO). NPY produces its effects by activating NPY receptors, of which there are at least five types (YI through Y2). NPY receptors are GPCRs. NPY can modify its own release and that of the other transmitters via presynaptic receptors of the Y2 type. NPY is removed from the synapse by metabolic breakdown by peptidases. ATP produces its effects by activating P2X receptors or P2Y receptors. P2X receptors are ligand-gated ion channels; P2Y receptors are GPCRs. There are multiple subtypes of both P2X and P2Y receptors. As with the other cotransmitters, ATP can act prejunctionally to modify its own release via receptors for ATP or via its metabolic breakdown to adenosine that acts on P1 (adenosine) receptors. ATP is cleared from the synapse primarily by releasable nucleotidases (rNTPase) and by cell-fixed ectonucleotidases.
MECHANISM OF ACTION OF α-ADRENERGIC RECEPTOR AGONISTS AND ANTAGONISTS
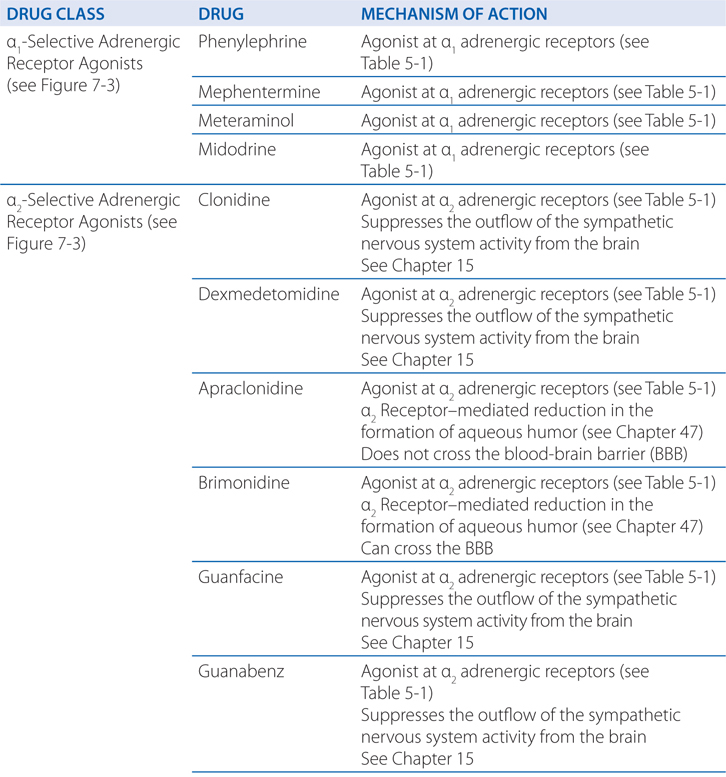
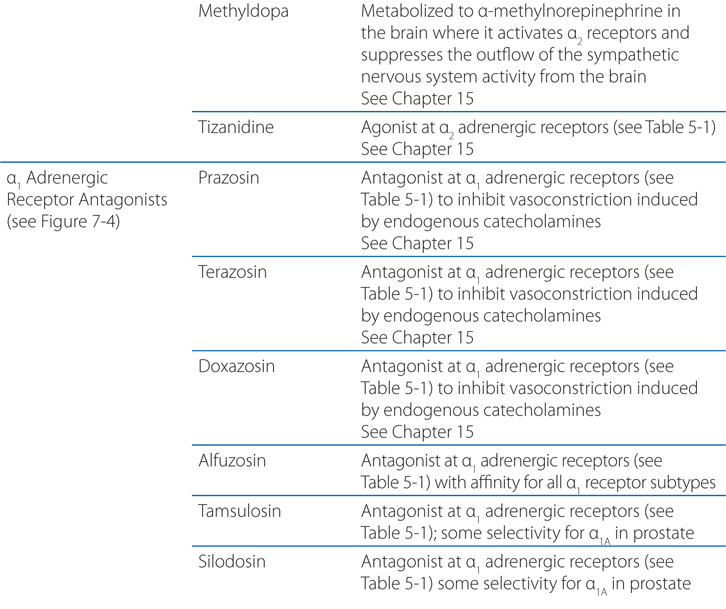
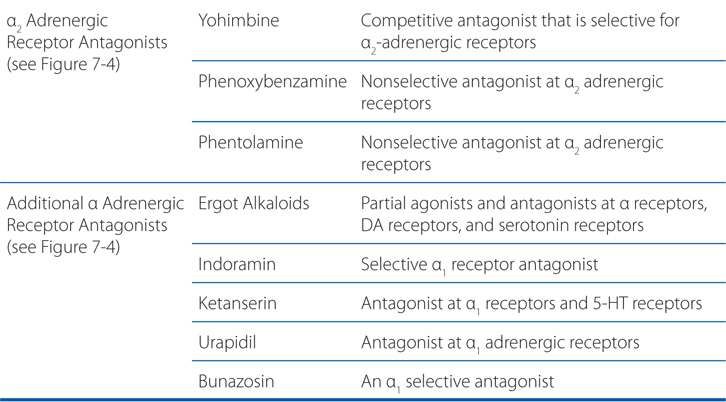
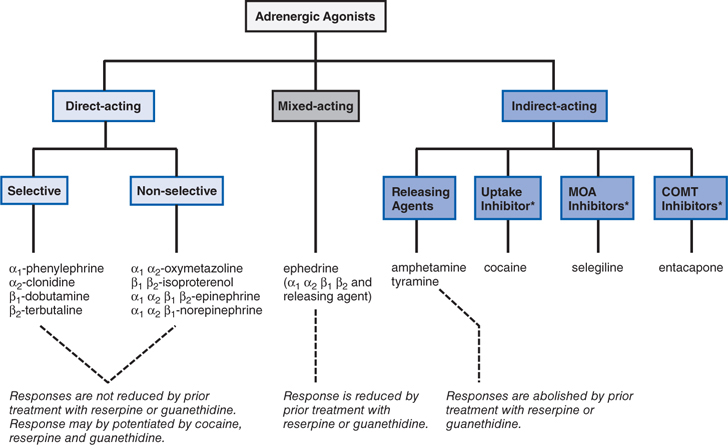
FIGURE 7-3 Classification of adrenergic receptor agonists (sympathomimetic amines) or drugs that produce sympathomimetic-like effects. For each category, a prototypical drug is shown. (*Not actually sympathetic drugs but produce sympathomimetic-like effects.)
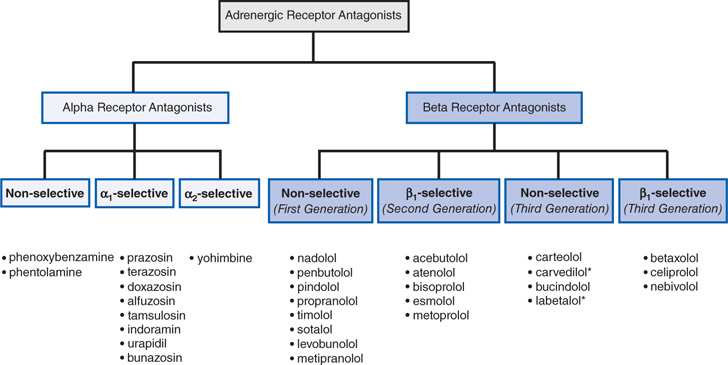
FIGURE 7-4 Classification of adrenergic receptor antagonists. Drugs marked by an asterisk (*) also block α1 receptors.
MECHANISM OF ACTION OF MISCELLANEOUS SYMPATHOMIMETIC AGONISTS
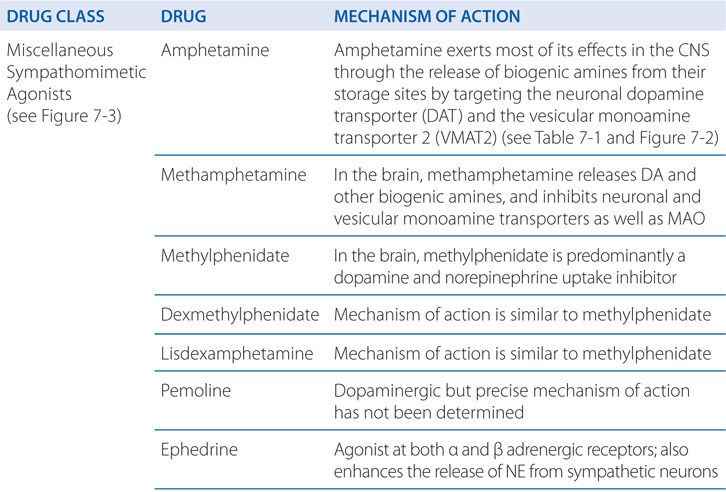

TABLE 7-1 Characteristics of Plasma Membrane Transporters for Endogenous Catecholamines
A 56-year-old woman is being treated with prazosin for her elevated blood pressure.
a. What class of drug is prazosin and how is it used to treat hypertension?
The major effects of prazosin result from its blockade of α1 receptors in arterioles and veins (see Figure 7-4). This leads to a fall in peripheral vascular resistance and in venous return to the heart. Unlike other vasodilating drugs, administration of prazosin usually does not increase heart rate (see answer to b below). In addition, prazosin decreases cardiac preload and thus has little tendency to increase cardiac output and rate, in contrast to vasodilators such as hydralazine that have minimal dilatory effects on veins (see Chapter 15).
MECHANISMS BY WHICH INDIRECT-ACTING SYMPATHOMIMETIC DRUGS INCREASE NE AND E TO STIMULATE ADRENERGIC RECEPTORS
• Release or displace NE from sympathetic nerve varicosities
• Block the transport of NE into sympathetic nerve terminals
• Block the catecholamine metabolizing enzymes MAO and COMT
b. What is prazosin’s mechanism of action?
Blockade of α1 adrenergic receptors inhibits vasoconstriction induced by endogenous catecholamines; vasodilation may occur in both arteriolar resistance vessels and in veins. The result is a fall in blood pressure due to decreased peripheral resistance. The magnitude of such effects depends on the activity of the sympathetic nervous system at the time the antagonist is administered, and thus is less in supine than in upright subjects and is particularly marked if there is hypovolemia. For most α receptor antagonists, the fall in blood pressure is opposed by baroreceptor reflexes that cause increases in heart rate as well as fluid retention. Prazosin appears to depress baroreflex function in hypertensive patients.
c. What are the major side effects of prazosin that this patient should be aware of?
A major potential adverse effect of prazosin and its congeners is the first-dose effect; marked postural hypotension and syncope sometimes are seen 30 to 90 minutes after an initial dose of prazosin. Syncopal episodes also have occurred with a rapid increase in dosage or with the addition of a second antihypertensive drug to the regimen of a patient who already is taking a large dose of prazosin. It is recommended that patients take their first dose immediately prior to bedtime.
A 48-year-old man with the diagnoses of essential hypertension is being treated with clonidine.
a. What type of drug is clonidine?
Clonidine, an α2-selective adrenergic agonist (see Figure 7-3), is used primarily for the treatment of systemic hypertension (see Chapter 15). Clonidine’s efficacy as an antihypertensive agent is somewhat surprising, because many blood vessels contain postsynaptic α2 adrenergic receptors that promote vasoconstriction. Indeed, clonidine, the prototypic α2 agonist, was initially developed as a vasoconstricting nasal decongestant.
b. What is clonidine’s mechanism of action?
Clonidine’s capacity to lower blood pressure results from activation of α2 receptors in the cardiovascular control centers of the CNS; such activation suppresses the outflow of sympathetic nervous system activity from the brain. Although the exact mechanism by which clonidine lowers blood pressure is not completely understood, the effect appears to result, at least in part, from activation of α2 receptors in the lower brainstem region.
c. What side effects of clonidine should this patient be warned about?
The major adverse effects of clonidine are dry mouth and sedation. These responses occur in at least 50% of patients and may require drug discontinuation. However, they may diminish in intensity after several weeks of therapy. Sexual dysfunction also may occur. Marked bradycardia is observed in some patients.
Withdrawal reactions follow abrupt discontinuation of long-term therapy with clonidine in some hypertensive patients.
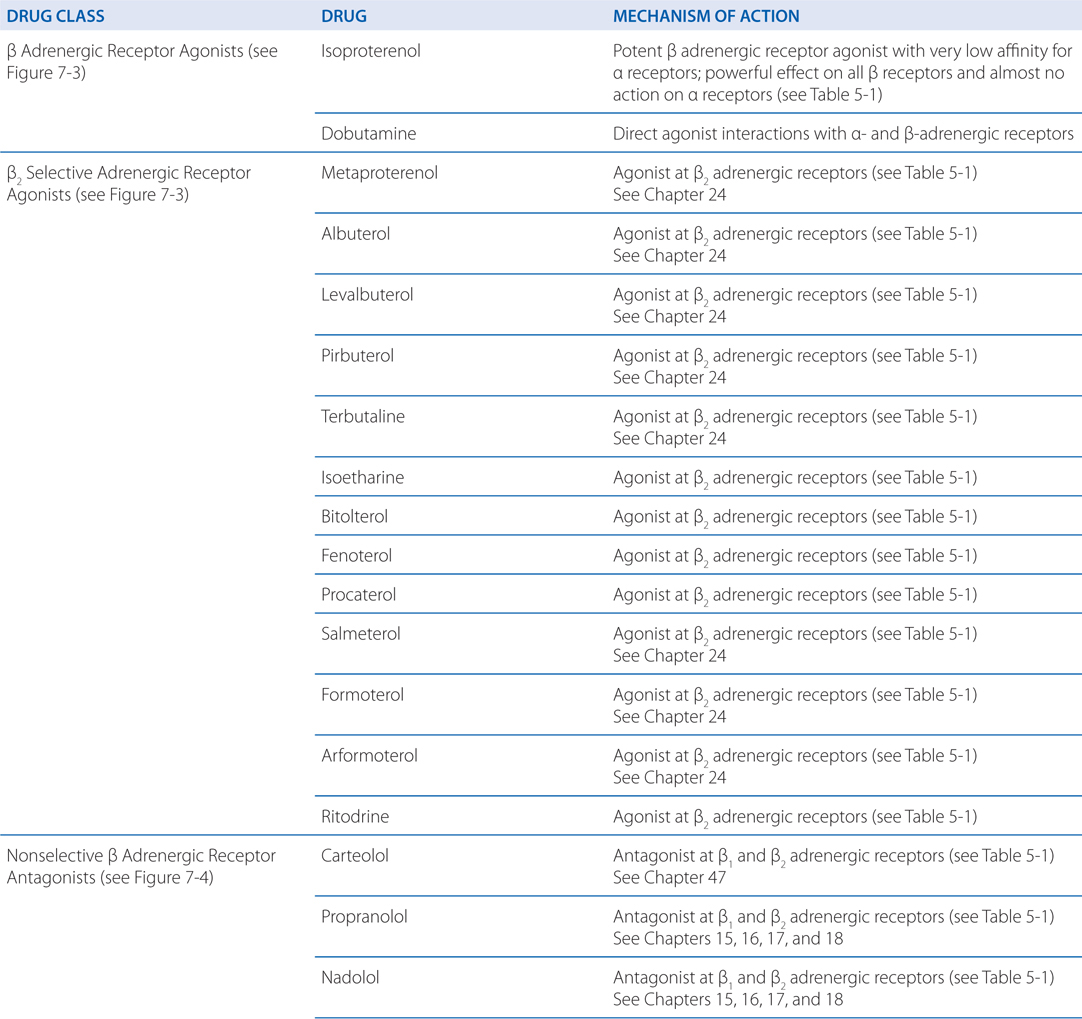
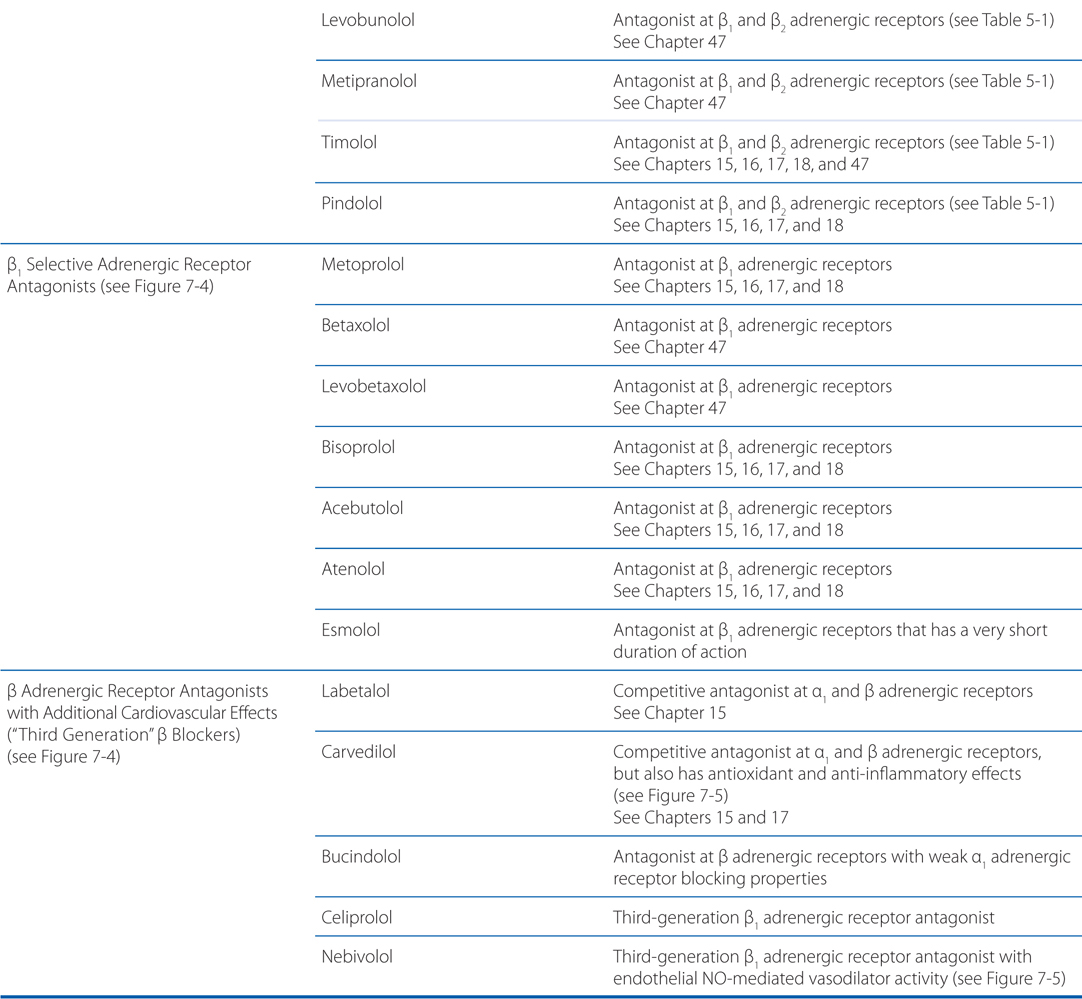
MECHANISM OF ACTION OF β ADRENERGIC RECEPTOR AGONISTS AND ANTAGONISTS
A 62-year-old woman with chronic obstructive airway disease is being treated with albuterol.
a. What kind of drug is albuterol?
Albuterolol, a selective β2 receptor agonist (see Figure 7-3), is administered either by inhalation or orally for the symptomatic relief of bronchospasm. Some of the major adverse effects of β receptor agonists in the treatment of asthma or obstructive pulmonary disease (COPD) are caused by stimulation of β1 receptors in the heart. Accordingly, drugs with preferential affinity for β2 receptors compared with β1 receptors have been developed. However, this selectivity is relative, not absolute, and is lost at high concentrations of these drugs.
b. How does albuterol affect airways so that this patient might expect improvement?
In the treatment of asthma and COPD, β receptor agonists are used to activate pulmonary receptors that relax bronchial smooth muscle and decrease airway resistance. Although this action appears to be a major therapeutic effect of these drugs in patients with asthma, evidence suggests that β receptor agonists also may suppress the release of leukotrienes and histamine from mast cells in lung tissue, enhance mucociliary function, decrease microvascular permeability, and possibly inhibit phospholipase A2. See Chapter 24 for a detailed discussion of the treatment of pulmonary disease with β2 receptor agonists.
c. What adverse effects should this patient be alerted to?
The major adverse effects of β receptor agonists occur as a result of excessive activation of β receptors. Tachycardia is a common adverse effect of systemically administered β receptor agonists. Stimulation of heart rate occurs primarily by means of β1 receptors. Patients with underlying cardiovascular disease are particularly at risk for significant reactions. However, the likelihood of adverse effects can be greatly decreased in patients with lung disease by administering the drug by inhalation rather than orally or parenterally.
The risk of adverse cardiovascular effects also is increased in patients who are receiving MAO inhibitors. In general, at least 2 weeks should elapse between the use of MAO inhibitors and administration of β2 agonists or other sympathomimetics.
Tremor is a relatively common adverse effect of the β2-selective receptor agonists. Tolerance generally develops to this effect. Feelings of restlessness, apprehension, and anxiety may limit therapy with these drugs, particularly oral or parenteral administration.
A 51-year-old man has suffered a myocardial infarction. In the intensive care unit, he is started on the β blocker, metoprolol.
a. Why is this patient started on a β blocker soon after the onset of his myocardial infarction?
A great deal of interest has focused on the use of β receptor antagonists in the treatment of acute myocardial infarction and in the prevention of recurrences for those who have survived an initial attack. Numerous trials have shown that β receptor antagonists administered during the early phases of acute myocardial infarction and continued long-term may decrease mortality by ~25%.
b. What is the mechanism of action of metoprolol?
The pharmacodynamics and pharmacokinetic parameters of β receptor antagonists are shown in Table 7-2. The precise mechanism by which β receptor antagonists decrease mortality and morbidity after a myocardial infarction is not known, but the favorable effects may stem from decreased myocardial oxygen demand, redistribution of myocardial blood flow, and antiarrhythmic actions. There is likely much less benefit if β adrenergic receptor antagonists are administered for only a short time. In studies of secondary prevention, the most extensive, favorable clinical trial data are available for propranolol, metoprolol, and timolol. In spite of these proven benefits, many patients with myocardial infarction do not receive a β adrenergic receptor antagonist.
TABLE 7-2 Pharmacological/Pharmacokinetic Properties of β Adrenergic Receptor Blocking Agents
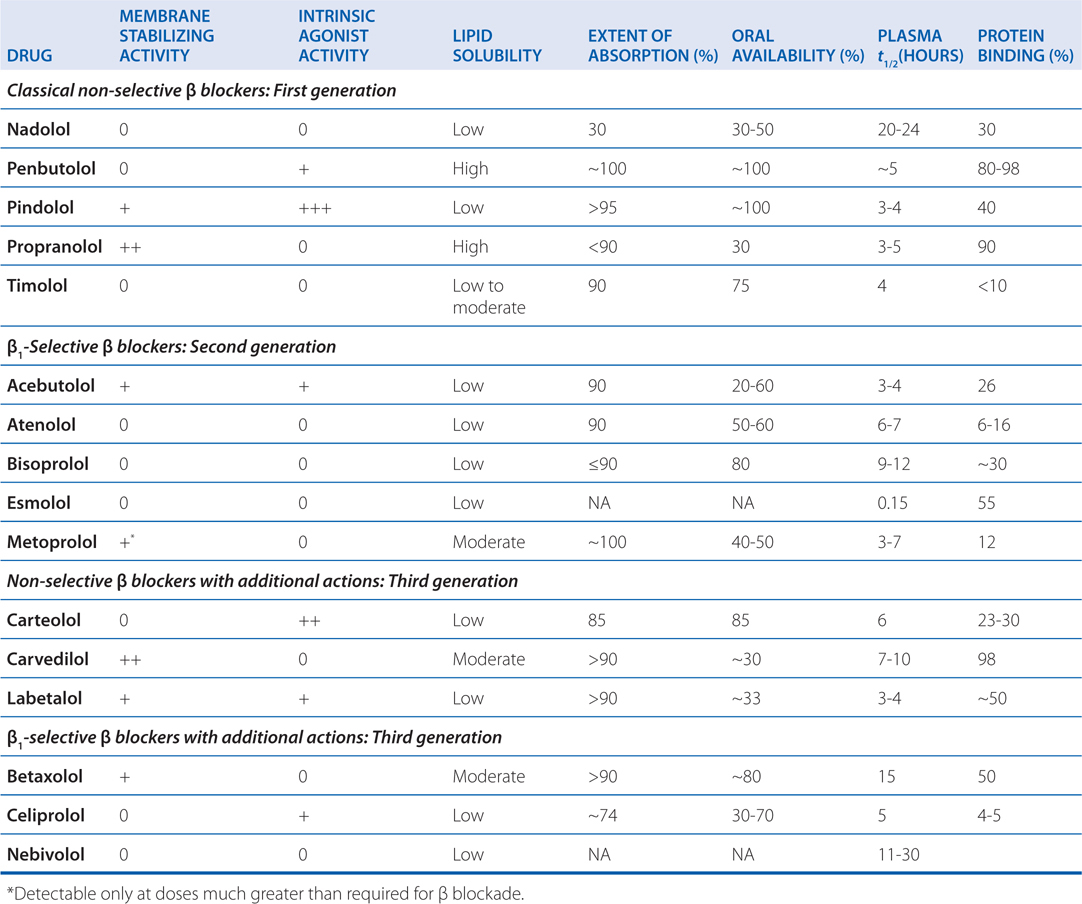
c. What are the adverse effects of β blockers?
The most common adverse effects of β receptor antagonists arise as pharmacological consequences of blockade of β receptors (see Table 5-1); serious adverse effects unrelated to β receptor blockade are rare. β Receptor blockade may cause or exacerbate heart failure in patients with decompensated heart failure, acute myocardial infarction, or cardiomegaly. Abrupt discontinuation of β receptor antagonists after long-term treatment can exacerbate angina and may increase the risk of sudden death.
A major adverse effect of β receptor antagonists is caused by blockade of β2 receptors in bronchial smooth muscle. Drugs with selectivity for β1 receptors or those with intrinsic sympathomimetic activity at β2 receptors seem less likely to induce bronchospasm. Since the selectivity of current β blockers for β1 receptors is modest, these drugs should be avoided if at all possible in patients with asthma.
The adverse effects of β receptor antagonists that are referable to the CNS may include fatigue, sleep disturbances (including insomnia and nightmares), and depression.
β Adrenergic blockade may blunt recognition of hypoglycemia by diabetic patients; it also may delay recovery from insulin-induced hypoglycemia. β Receptor antagonists should be used with great caution in patients with diabetes who are prone to hypoglycemic reactions; β1-selective agents may be preferable for these patients.
A 58-year-old woman has suffered from mild heart failure for 5 years. She has been treated with carvedilol. She recently changed to a different physician who questions the use of a β blocker to treat heart failure.
a. Why would her new physician question the use of a β blocker to treat heart failure?
It is a common clinical observation that administration of β receptor antagonists can worsen markedly or even precipitate congestive heart failure in compensated patients with multiple forms of heart disease, such as ischemic or congestive cardiomyopathy. Consequently, the hypothesis that β receptor antagonists might be efficacious in the long-term treatment of heart failure originally seemed counterintuitive to many physicians.
Carvedilol, metoprolol, and bisoprolol all have been shown to reduce the mortality rate in large cohorts of patients with stable chronic heart failure regardless of severity. Treatment of patients with congestive heart failure with β receptor antagonists should only be undertaken by physicians with experience in using these drugs for this purpose.
b. What is the mechanism of action of carvedilol for this indication?
A number of mechanisms have been proposed to play a role in the beneficial effects of β receptor antagonists in heart failure. Since chronic excess catecholamines may be toxic to the heart, especially through activation of β1 receptors, inhibition of the pathway may help preserve myocardial function. Also, antagonism of β receptors in the heart may attenuate cardiac remodeling, which ordinarily might have deleterious effects on cardiac function. Interestingly, activation of β receptors and elevation of cellular cyclic AMP may promote myocardial cell death by apoptosis. In addition, properties of certain β receptor antagonists that are due to other, unrelated properties of these drugs may be potentially important. For example, afterload reduction by drugs such as carvedilol may be clinically relevant (see Table 7-3 and Figure 7-5).
TABLE 7-3 Third Generation β Receptor Antagonists with Putative Additional Mechanisms of Vasodilation
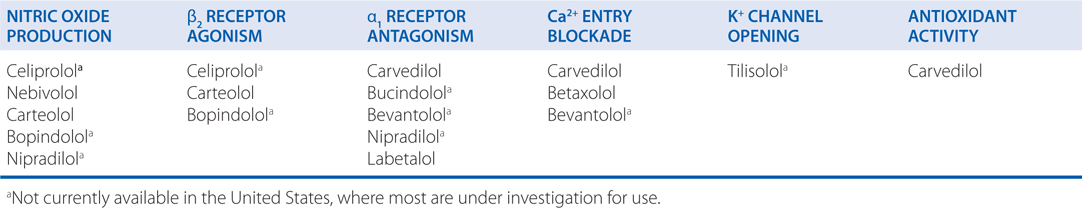
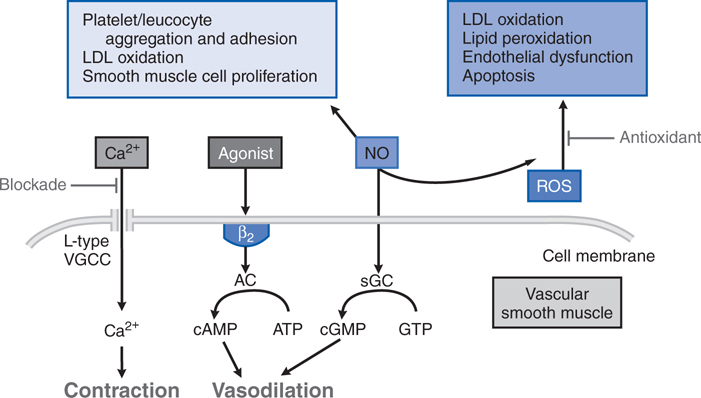
FIGURE 7-5 Mechanisms underlying actions of vasodilating β blockers in blood vessels. ROS, reactive oxygen species; sGC, soluble guanylyl cyclase; AC adenylyl cyclase; L-type VGCC, L-type voltage gated Ca2+ channel. (Modified with permission from Toda N. Vasodilating β-adrenoceptor blockers as cardiovascular therapeutics. Pharmacol Ther, 2003;100:215-234. Copyright © Elsevier.)
MECHANISM OF ACTION OF DRUGS ACTING AT SEROTINERGIC AND DOPAMINERGIC RECEPTORS
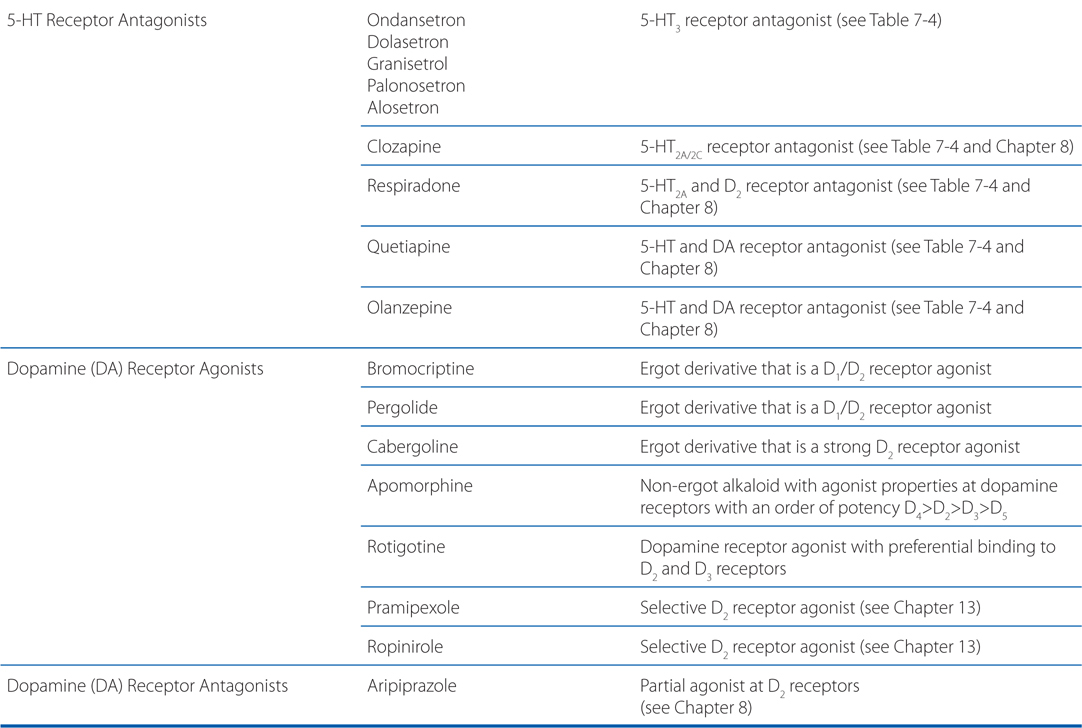
A 36-year-old woman has been going through a difficult divorce for the past year. Two weeks ago her father died. She now presents to her physician with moderate situational depression and is started on the selective serotonin reuptake inhibitor (SSRI) fluoxetine.
a. Why is a drug that modulates serotonin (5-HT) at nerve endings effective in treating depression?
The synthesis and inactivation of serotonin is shown in Figure 7-6 and the actions and clinical indications of serotonergic drugs are presented in Table 7-5. A multitude of brain functions are influenced by 5-HT, including sleep, cognition, sensory perception, motor activity, temperature regulation, nociception, mood, appetite, sexual behavior, and hormone secretion. All of the cloned 5-HT receptors are expressed in the brain, often in overlapping domains (see Table 7-4). Although patterns of 5-HT receptor expression in individual neurons have not been extensively defined, it is likely that multiple 5-HT receptor subtypes with similar or opposing actions are expressed in individual neurons, leading to a tremendous diversity of actions.
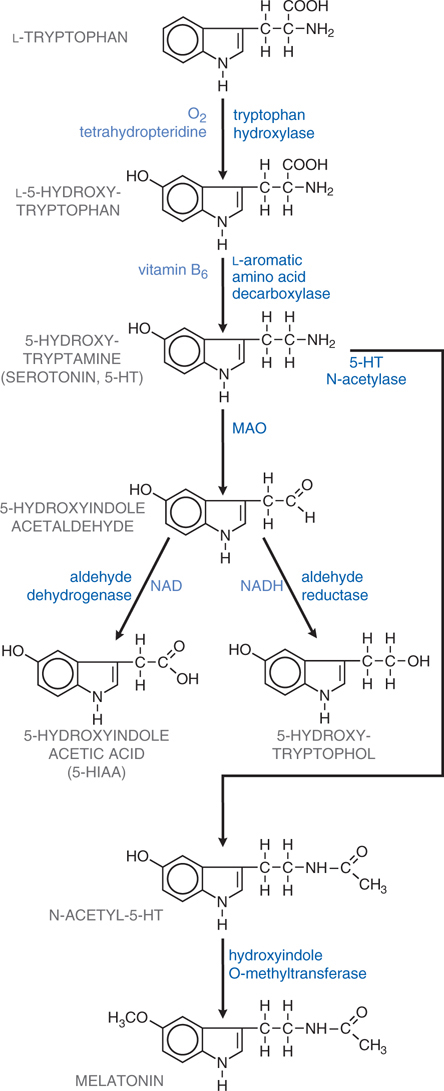
FIGURE 7-6 Synthesis and inactivation of serotonin. Enzymes and co-factors are shown in blue.
TABLE 7-4 Serotonin Receptor Subtypes
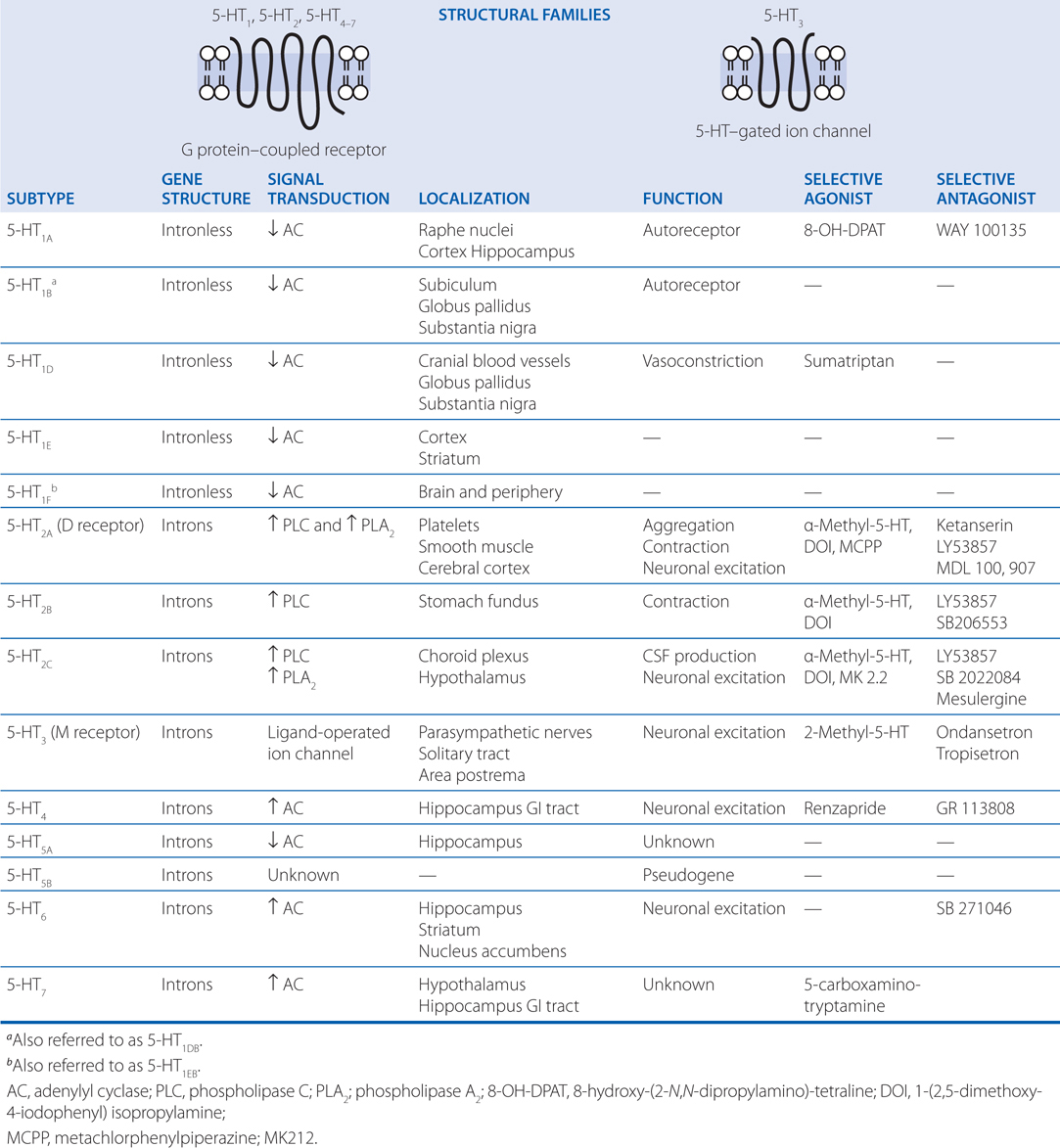
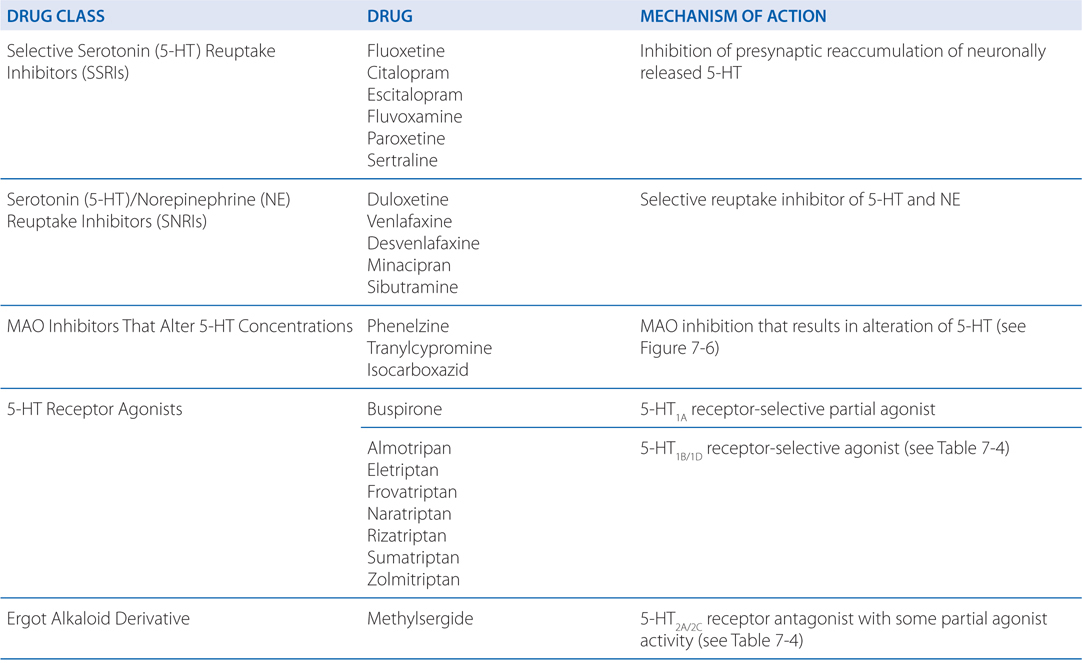
TABLE 7-5 Serotonergic Drugs: Primary Actions and Clinical Indications
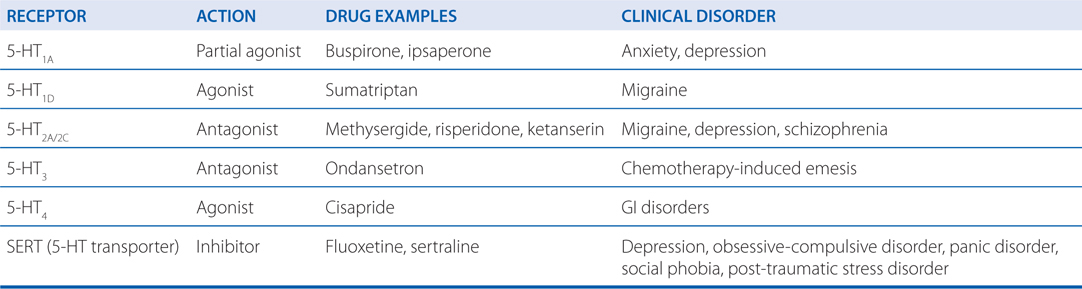
The effects of 5-HT–active drugs in anxiety and depressive disorders, like the effects of selective serotonin reuptake inhibitors (SSRIs), strongly suggest a role for 5-HT in the neurochemical mediation of these disorders.
EVIDENCE SUGGESTING THAT 5-HT IS A KEY MEDIATOR IN THE PATHOGENESIS OF MIGRAINE
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


