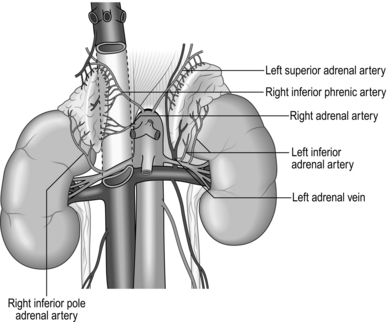
22 The normal adrenal gland weighs 3–5 g and measures 5x3x1 cm. Both left and right glands lie in the retroperitoneum within the perirenal (Gerota’s) fascia, with their posterior surface attached to the diaphragm. They receive arterial blood supply from several small branches of the inferior phrenic artery, aorta and renal artery. The adrenals are not symmetrical: the right is triangular in shape and its short, wide adrenal vein drains medially into the inferior vena cava; the left is more semilunar, with the vein draining downwards into the left renal vein. Each adrenal gland consists of medulla, secreting catecholamines (adrenaline (epinephrine), noradrenaline (norepinephrine) and dopamine), and cortex, secreting cortisol, aldosterone and adrenal sex hormones (Fig. 22.1). Lymphatic capillaries draining the cortex follow arteries while the medulla lymphatics follow veins. Before you embark on adrenalectomy, consider: 1. The functional status of the adrenal nodule, as this determines patient preparation before surgery and perioperative management. 2. Look at the size of the adrenal mass and its relation to surrounding organs. This determines your choice of incision and approach. You must review CT or MRI scans yourself and have them available in the operating theatre during surgery. 3. Decide whether you are dealing with malignant or benign pathology. 4. Find out whether this is a sporadic or familial problem. 5. Ensure that surgery is the best treatment and is really necessary. 1. Phaeochromocytomas arise from adrenal medulla and secrete adrenaline (epinephrine), noradrenaline (norepinephrine) and dopamine. It is difficult to predict their biological behaviour but 10% of them are malignant and 10% arise from chromaffin cells outside the adrenals (paragangliomas). Most of them are sporadic, but 20–30% are familial and may be bilateral. Patients present with headaches, sweating, hypertension and palpitations. Establish the biochemical diagnosis by measuring catecholamine and metanephrine levels in urine and plasma. CT, MRI and MIBG (meta-iodobenzyl-guanidine) scans are used to assess tumour location and size and to detect possible metastases. Carefully prepare all patients with phaeochromocytomas with α blockade and sometimes β blockade to minimize the risk of hypertensive crisis. During surgery avoid extensive manipulation of the tumour to prevent a sudden rise in blood pressure. You should work with an experienced anaesthetist who is able to control surges of blood pressure during dissection and hypotension after the removal of the tumour. 2. Primary hyperaldosteronism (Conn’s syndrome) is caused by excess of aldosterone produced by either a solitary adenoma or bilateral hyperplasia of the zona glomerulosa of the adrenal cortex. Patients present with hypertension and low potassium. Biochemical diagnosis is confirmed by an elevated plasma aldosterone concentration and suppressed plasma renin activity. Use CT or MRI scans to identify adrenal nodules, which are usually about 1 cm in size. Adrenal venous sampling is sometimes necessary to differentiate unilateral from bilateral disease. The aim of the treatment is to normalize aldosterone levels and prevent mortality and morbidity caused by hypertension, low potassium, cardiovascular and renal damage. Unilateral adenomas should be treated with adrenalectomy, which corrects hypokalaemia in 98% and improves hypertension in 90% of patients. Patients with bilateral hyperplasia should be treated with mineralocorticoid receptor antagonists (e.g. spironolactone), not surgery. 3. Hypercorticolism is caused by excess of cortisol. Cushing’s disease is caused by pituitary ACTH producing adenomas, which stimulates adrenal production of cortisol. Twenty-five percent of patients are not cured by transphenoidal pituitary surgery and require bilateral adrenalectomy. Cushing’s syndrome is caused by a cortisol-secreting adenoma or rarely adrenocortical carcinoma and is an indication for unilateral adrenalectomy. Patients present with central obesity, hypertension, diabetes and muscle weakness. They have impaired immunity and poor wound healing. Biochemical diagnosis is made by measuring plasma ACTH and urine cortisol and performing a dexamethasone suppression test. Before surgery hypercorticolism can be controlled with ketaconazole or metyrapone. After bilateral adrenalectomy patients need lifelong treatment with hydrocortisone and mineralocorticoids. The contralateral gland is often suppressed after unilateral adrenalectomy and these patients also need treatment with hydrocortisone for many months. 4. Adrenocortical carcinoma is a rare but highly malignant tumour with poor prognosis. 60% of adrenal cancers are hormonally active and can cause Cushing’s or virilizing syndromes. Most of these tumours are large at diagnosis and an open approach should be used to resect them. Surgery often needs to be extensive with en bloc removal of adrenal and surrounding organs combined with lymphadenopathy. Tumour spillage must be avoided at all cost to prevent local recurrence. 5. Adrenal ‘incidentaloma’ is a mass discovered by chance during radiological investigations performed for other indications. They are common, with a prevalence of 4–6%. Most of them (70%) are non-secreting adenomas, but about 16% are hyperfunctioning and cause subclinical Cushing’s, Conn’s or phaeochromocytoma syndromes. Incidentalomas larger than 4–5 cm may represent malignant pathology. Hyperfunctioning and large incidentalomas suspicious of being malignant should be resected. 1. With the correct diagnosis established and the perioperative plan discussed with an endocrinologist and implemented, you can now plan your surgical procedure. Depending on pathology you must make a decision:
Adrenalectomy
INTRODUCTION
ANATOMY
Appraise
Indications for adrenalectomy
Prepare
 Whether to perform unilateral or bilateral adrenalectomy
Whether to perform unilateral or bilateral adrenalectomy
 Which access is best (anterior, lateral, posterior)
Which access is best (anterior, lateral, posterior)
![]()
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree