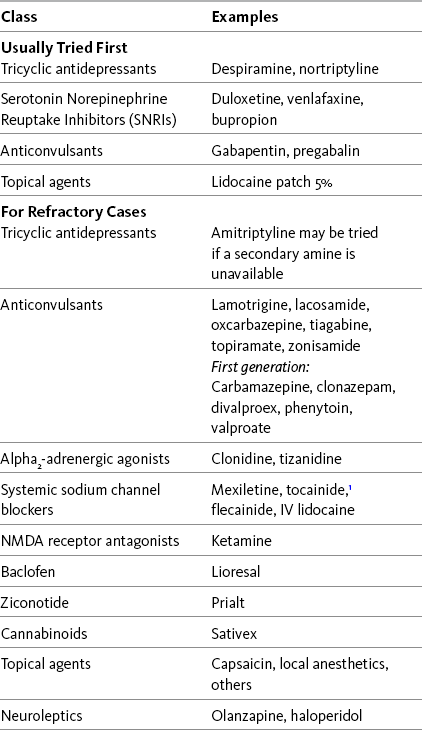
Chapter 23 SOME of the adjuvant analgesic classes are conventionally used solely for persistent neuropathic pain. The drugs in these classes, combined with the drugs in classes subsumed under the category of multipurpose analgesics, offer a very large group of individual agents that might be useful for pains of this type. Antidepressants and anticonvulsants are the first-line adjuvant analgesics for a wide variety of neuropathic pain syndromes. The multipurpose antidepressants were discussed earlier in Chapter 22, and the anticonvulsants will be discussed in detail here. Refractory neuropathic pain, which has not responded to these first-line approaches, may be considered for trials of the other so-called multipurpose drugs, or other agents classified as drugs used conventionally for neuropathic pain. Other adjuvant agents used for refractory neuropathic pain include sodium channel blockers, several topical agents, gamma aminobutyric acid (GABA) agonists (baclofen [Lioresal]), N-methyl-d-aspartate (NMDA) receptor antagonists (e.g., dextromethorphan and ketamine), and the relatively new intrathecal drug, ziconotide (Prialt). In addition to persistent pain, certain adjuvant agents are used to manage the neuropathic component of some types of acute pain and for the purpose of preventing persistent neuropathic pain, such as persistent neuropathic postsurgical pain. These are discussed later in this section. Recent systematic reviews, some with evidence-based guidelines for drug selection, provide information about a range of therapies used for neuropathic pain (Finnerup, Otto, McQuay, et al., 2005; Saarto, Wiffen, 2007; Kroenke, Krebs, Bair, 2009). Table 23-1 lists many of these drugs, and Table V-1 at the end of Section V provides dosing guidelines and other characteristics of many of the adjuvant analgesics for the treatment of neuropathic pain. See also Section II for assessment of neuropathic pain. Guidelines Table 23-1 Adjuvant Analgesics for Neuropathic Pain IV, Intravenous; NMDA, N-methyl-d-aspartate. 1No longer marketed in the United States. From Pasero, C., & McCaffery, M. Pain assessment and pharmacologic management, p. 654, St. Louis, Mosby. Pasero C, McCaffery M. May be duplicated for use in clinical practice. Following is a discussion of several of the “newer” second-generation anticonvulsants. See Chapter 26 for their use in acute pain treatment. Gabapentin (Neurontin) has been demonstrated to be analgesic in many types of neuropathic pain, some other types of persistent pain, and acute perioperative pain (Knotkova, Pappagallo, 2007; Kong, Irwin, 2007; Seib, Paul, 2006; Tiippana, Hamunen, Kontinen, et al., 2007; Wiffen, McQuay, Rees, et al., 2005). (See Chapter 26 for perioperative use of gabapentin.) For example, a large randomized, placebo-controlled study of 305 patients with diverse neuropathic pain syndromes, including postherpetic neuralgia, complex regional pain syndrome (CRPS), central pain, and persistent postsurgical pain, found that gabapentin in doses up to 2400 mg were well-tolerated and improved pain intensity by 21% compared with 14% with placebo (Serpell, Neuropathic Pain Study Group, 2002). Improvements were noted in patient-reported quality of life and functional indicators as well. Not all studies in neuropathic pain have yielded positive results. In an 8-week 3-phase crossover trial (N = 38) that compared gabapentin, amitriptyline, and diphenhydramine (Benadryl) for spinal cord injury–related pain, gabapentin was no more effective than diphenhydramine in patients who had the highest baseline pain scores; amitriptyline was more effective than diphenhydramine (Rintala, Holmes, Courtade, et al., 2007) (see Chapter 31 for more on antihistamines). • Postherpetic neuralgia (Backonja, Glanzman, 2003; Chou, Carson, Chan, 2009; Dubinsky, Kabbani, El-Chami, et al., 2004; Mao, Chen, 2000a; Rosenberg, Harrell, Ristic, et al., 1997; Rice, Maton, Postherpetic Neuralgia Study Group, 2001; Rosner, Rubin, Kestenbaum, 1996; Rowbotham, Harden, Stacey, et al., 1998; Serpell, Neuropathic Pain Study Group, 2002) • Painful diabetic neuropathy (Argoff, Backonja, Belgrade, et al., 2006; Backonja, Beydoun, Edwards, et al., 1998; Backonja, Glanzman, 2003; Boulton, Vinik, Arezzo, et al., 2005; Chou, Carson, Chan, 2009; Dallocchio, Buffa, Mazzarello, et al., 2000; Duby, Campbell, Setter, et al., 2004; Gilron, Bailey, Tu, et al., 2005; Hemstreet, Lapointe, 2001; Jensen, Larson, 2001; Mao, Chen, 2000a; Serpell, Neuropathic Pain Study Group, 2002; Veves, Backonja, Malik, 2008) • Fibromyalgia (Arnold, Goldenberg, Stanford, et al., 2007) (see also Hauser, Thieme, Turk, 2009) • Neuropathic cancer pain (Caraceni, Zecca, Martini, et al., 1999; Keskinbora, Pekel, Aydinli, 2007) • Chemotherapy-induced pain (Mao, Chen, 2000a; Tsavaris, Kopterides, Kosmas, et al., 2008) • Central pain from spinal cord injury (To, Lim, Hill, et al., 2002; (Levendoglu, Ogun, Ozerbil, et al., 2004) • Central poststroke pain (Frese, Husstedt, Ringelstein, et al., 2006; Kumar, Kalita, Kumar, et al., 2009; Serpell, Neuropathic Pain Study Group, 2002) • Multiple sclerosis (Mao, Chen, 2000a) • Phantom limb pain (Bone, Critchley, Buggy, 2002; Serpell, Neuropathic Pain Study Group, 2002) • CRPS (Backonja, Glanzman, 2003; Mao, Chen, 2000a; Mellick, Mellick, 1995; Serpell, Neuropathic Pain Study Group, 2002) • Radiculopathy (Serpell, Neuropathic Pain Study Group, 2002) • HIV-related neuropathy (Rosner, Rubin, Kestenbaum, 1996; Hahn, Arendt, Braun, et al., 2004) • Spinal stenosis (Yaksi, Ozgonenel, Ozgonenel, 2007) • Atypical facial pain and trigeminal neuralgia (Mao, Chen, 2000a; Serpell, Neuropathic Pain Study Group, 2002) • Cluster and migraine headache (Kaniecki, 2008; Mathew, 2001; Tay, Ngan Kee, Chung, 2001) • Neuroma of peripheral nerve (Serpell, Neuropathic Pain Study Group, 2002) • Persistent neuropathic postsurgical pain syndromes (e.g., postmastectomy, postthoractomy, post inguinal hernia, cholecystectomy) (Backonja, Glanzman, 2003; Mao, Chen, 2000a; Pandey, Patra, Pant, et al., 2006; Serpell, Neuropathic Pain Study Group, 2002; Tiippana, Hamunen, Kontinen, et al., 2007) • Persistent back pain (Backonja, Glanzman, 2003; Chou, Qaeem, Snow, et al., 2007; Serpell, Neuropathic Pain Study Group, 2002) • Persistent masticatory muscle pain (Kimos, Biggs, Mah, et al., 2007) • Guillain-Barré syndrome (Mao, Chen, 2000a; Pandey, Bose, Garg, et al., 2002) The onset of pregabalin analgesia is approximately 25 minutes (Hill, Balkenohl, Thomas, et al., 2001), compared with 1 to 3 hours for gabapentin (Twycross, Wilcock, Charlesworth, et al., 2003). This faster onset of analgesic action may be clinically relevant in some cases (Blommel, Blommel, 2007). Equally important, pregabalin can be more rapidly titrated to the typical effective dose range than gabapentin. The time to effective dose for pregabalin may be as brief as 1 to 2 days (Gajraj, 2007; Portenoy, Murphy, Young, et al., 2006), compared to approximately 9 days for gabapentin (Gajraj, 2007). (See Chapter 26 for the perioperative use of pregabalin.) Recent evidence-based guidelines indicate that pregabalin (or gabapentin) should be considered the first-line drug for the treatment of postherpetic neuralgia, painful diabetic neuropathy, and other neuropathic pains, unless a co-morbid depression suggests that an analgesic antidepressant should be tried first (Argoff, Backonja, Belgrade, et al., 2006; Dworkin, O’Connor, Backonja, et al., 2007) (see Chapter 22). This conclusion gains support from the consistent results observed in randomized controlled trials. • Painful diabetic neuropathy (Argoff, Backonja, Belgrade, et al., 2006; Baron, Brunnmuller, Brasser, et al., 2007; Boulton, Vinik, Arezzo, et al., 2005; Frampton, Scott, 2004; Frank, Cousins, 2008; Freeman, Durso-Decruz, Emir, 2008; Freynhagen, Strojek, Griesing, et al., 2005; Freynhagen, Grond, Schupfer, et al., 2007; Richter, Portenoy, Sharma, et al., 2005; Rosenstock, Tuchman, LaMoreaux, et al., 2004; Tolle, Freynhagen, Versavel et al., 2008; Veves, Backonja, Malik, 2008) • Fibromyalgia (Arnold, Crofford, Martin, et al., 2007; Arnold, Russell, Diri, et al., 2008; Crofford, Rowbotham, Mease, et al., 2005) (see also Hauser, Thieme, Turk, 2009.) • Postherpetic neuralgia (Baron, Brunnmuller, Brasser, et al., 2007; Dubinsky, Kabbani, El-Chami, et al., 2004; Dworkin, Corbin, Young, et al., 2003; Frampton, Foster, 2005; Freynhagen, Strojek, Griesing, et al., 2005; Rowbotham, Stacey, Phillips, et al., 2007; Stacey, Barrett, Whalen, et al., 2008; van Seventer, Feister, Young, et al., 2006) • Central pain from brain or spinal cord injury (Siddall, Cousins, Otte, et al., 2006; Vranken, Dijkgraaf, Kruis, et al., 2008) • Trigeminal neuralgia (Obermann, Yoon, Sensen, et al., 2008) • Glossopharyngeal neuralgia (Guido, Specchio, 2006) • Restless leg syndrome with or without neuropathic pain (Sommer, Bachmann, Liebetanz, et al., 2007). Clonazepam (Klonopin) is a benzodiazepine and has been used primarily as an anticonvulsant. It has been suggested to be analgesic in the lancinating pain associated with phantom limb pain, neuropathic cancer-related pain, and myofascial pain (Bartusch, Sanders, D’Alessio, et al., 1996; Hugel, Ellershaw, Dickman, 2003; Fishbain, Cutler, Rosomoff, et al., 2000). The supporting evidence is very limited, however, and its use may be more related to its potential to help co-morbid anxiety than to its established analgesic efficacy. Given its long half-life, the potential for accumulation with repeated dosing must be recognized, and it must be used very cautiously in patients predisposed to adverse effects, including the cognitively impaired (particularly older adults) and patients with sleep apnea syndrome or advanced cardiopulmonary disease. (See clonazepam patient medication information, Form V-3 on pp. 763-764, at the end of Section V.) One of the mechanisms is blockade of presynaptic voltage-gated ion channels, which prevents the generation of spontaneous ectopic discharges (Beydoun, Backonja, 2003; Taylor, 2009) (see Section I and Figure I-2, B on pp. 4-5). Some anticonvulsants (e.g., carbamazepine, felbamate, lamotrigine, oxcarbazepine, phenytoin, topiramate, and zonisamide) relieve pain, in part, by prolonging the recovery phase of sodium channels after their activation (Gilron, 2006; Soderpalm, 2002). Some (e.g., carbamazepine, felbamate, gabapentin, lamotrigine, pregabalin, valproic acid, and zonisamide) bind to presynaptic voltage-gated calcium channels and inhibit calcium influx and the release of excitatory neurotransmitters from primary afferent nerve fibers (Dickenson, Matthews, Suzuki, 2002; Gajraj, 2007; Gilron, 2006; Taylor, 2009; Tiippana, Hamunen, Kontinen, et al., 2007). Pregabalin has an even higher calcium-channel affinity than gabapentin and has no effect on sodium channels (Gilron, Watson, Cahill, et al., 2006; Mao, Chen, 2000a; Nicholson, 2000). Recent research confirmed that gabapentin had no effect on transient sodium currents but inhibited persistent sodium currents in a dose-dependent way (Yang, Wang, Chen, et al., 2009). The clinical significance of these findings is unknown. Other mechanisms also may be important. Inhibition of glutamate, an excitatory neurotransmitter that promotes the transmission of pain through increased activity at the NMDA receptor and other receptors, may be relevant for some drugs (Jensen, 2002; Soderpalm, 2002) (see Section I and Figure I-2, B on pp. 4-5). Others may indirectly or directly augment inhibitory GABAergic neurotransmission (Gilron, 2006; Soderpalm, 2002). For example, gabapentin is a GABA analogue, and although it does not bind to GABA receptors or modulate GABA reuptake, it does enhance overall GABA-mediated inhibitory tone (Dickenson, Matthews, Suzuki, 2002; Jensen, 2002; Mao, Chen, 2000). Gabapentin and pregabalin share a specific high affinity drug binding site (calcium channel α2-δ ligands) localized at synapses and sufficient in amount to account for their analgesic action (Taylor, 2009). Although there have been case reports of hepatotoxicity with gabapentin (Richardson, Williams, Kingham, 2002), they are rare. Neither gabapentin nor pregabalin undergo hepatic metabolism, a positive effect of which is a minimal risk of drug-drug interactions (Frank, Cousins, 2008). The new anticonvulsant, lacosamide, produces low or no inhibition of cytochrome P450 isoenzymes and also may have a reduced risk of interactions (Kropeit, Scharfenecker, Schiltmeyer, et al., 2006). Other anticonvulsants inhibit various isoenzymes of the cytochrome P450 enzyme system, which can result in drug-drug interactions (Virani, Mailis, Shapiro, et al., 1997) (see Chapter 11, for more on the cytochrome P450 enzyme system). Each anticonvulsant has a unique profile (LaRoche, 2007), and it is important for the clinician to become familiar with the potential for drug-drug interactions associated with the particular anticonvulsant being administered. Patients must be told to report adverse effects promptly, and in some cases serum drug concentrations should be closely monitored to prevent toxicity, such as in patients who take multiple other medications and those who take anticonvulsants during chemotherapy (Yap, Chui, Chan, 2008). Following is a more detailed discussion of adverse effects associated with selected anticonvulsants. As mentioned, a major benefit of gabapentin is that it is not hepatically metabolized. This results in minimal drug-drug interactions and hepatic adverse effects. The drug is excreted entirely by the renal system. Gabapentin toxicity was reported in a case describing its use during an episode of acute renal failure (Miller, Price, 2009). The importance of recognizing the need to adjust the dose downward during acute illness, particularly when there is a decline in renal clearance, must be recognized. (See gabapentin patient medication information, Form V-7 on pp. 771-772, at the end of Section V.) Also, like gabapentin, pregabalin does not undergo hepatic metabolism and has no reported drug interactions of concern (Frank, Cousins, 2008). Additive pharmacodynamic effects of the type that may occur whenever two or more centrally-acting drugs are taken also occurs during pregabalin dosing; patients may report effects on cognitive and gross-motor function when the drug is co-administered with other drugs or alcohol (Blommel, Blommel, 2007). Being a relatively new drug, evaluation of the impact of its adverse effects in older patients requires further research and clinical experience (Guay, 2005). (See pregabalin patient medication information, Form V-11 on pp. 779-780, at the end of Section V.)
Adjuvant Analgesics for Persistent (Chronic) Neuropathic Pain
Anticonvulsant Drugs
“Newer” Second-Generation Anticonvulsants
Gabapentin
Pregabalin
First-Generation Anticonvulsants
Clonazepam
Mechanism of Action
Adverse Effects
Gabapentin
Pregabalin
![]()
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


Basicmedical Key
Fastest Basicmedical Insight Engine
