, Johann Emmanuel2 and Brendon J. Coventry3
(1)
Department of Anaesthesia and Pain Management, Pain Management Research Institute, Royal North Shore Hospital, University of Sydney, St Leonards, NSW, Australia
(2)
Pain Management Research Institute, Royal North Shore Hospital, University of Sydney, St Leonards, NSW, Australia
(3)
Royal Adelaide Hospital, University of Adelaide, L5 Eleanor Harrald Building, North Terrace, 5000 Adelaide, SA, Australia
Abstract
Tissue trauma, including from acute trauma, acute surgical pathology, and surgical intervention, typically results in pain as a natural consequence. With an improved understanding of pain genesis, pathways, and pharmacology, much of the pain from surgery can be can usually be minimized or alleviated with adequate planning, consideration, and utilization of a multimodality approach. The aim of this chapter is to explain the principles of acute and chronic pain in the perioperative period and to assist with a better understanding for more effective pain control in surgical practice.
Overview
Most patients anticipated some degree of postoperative pain as an associated feature of surgery and a consequence of performing a surgical incision. However, with better anesthesia and analgesia, many patients have now come to expect less pain than previous generations endured. This in itself is an important consideration, as anticipation of the extent of the pain may modify the actual perception of pain, and additionally, patient “satisfaction” with medical/surgical treatment may be heavily contingent upon their prior expectation of the intensity of pain they might suffer when agreeing during “informed consent” to a procedure. Realistically, however, some pain is usually expected, by most patients, with most surgical procedures. In effect, with much improved analgesia (thankfully), we also inadvertently “raise the bar” of patient and community expectation for “pain-free” surgical and anesthetic care. Despite the enormous advances in anesthesia and analgesia over the last 50 years, some postoperative pain is still experienced by many patients, and the overall aim is therefore to make the patient as comfortable and as least distressed by the pain, as possible. Indeed, the so-called “breakthrough” pain is important for the surgical diagnosis of postoperative complications, such as vascular compartment syndrome in a limb or following an undetected gastrointestinal perforation or an anastomotic leak. Not infrequently, pain precedes surgery, especially with acute surgical conditions or following trauma, and so the postoperative pain potentially arising from surgery is often a mix of preoperative pain, any intraoperative pain, and the postoperative pain induced by surgical intervention. Indeed, for many patients in pain prior to surgery, postoperative pain is a lesser and a much preferable option. Postoperative pain as a pathophysiological response is therefore important but must be managed appropriately to reduce the amount of distress and discomfort to acceptable levels for the patient and in a safe manner. This chapter focuses on some of the underlying mechanisms, types of pain experienced by patients, pain associated with complication following surgery, and its possible management.
Introduction
International Association for the Study of Pain (IASP) has defined pain as “an unpleasant sensory and emotional experience associated with actual or potential tissue damage, or described in terms of such damage” (Merskey 1979). An inability to verbally communicate does not mean that an individual is not experiencing pain and does not negate the need of suitable pain-relieving treatment. This is important for certain situations or conditions including intensive care, coma, during anesthesia, dementia, retardation, dysarthria, and in pediatric patients. Noxious stimuli in the absence of tissue damage usually result in temporary acute pain, as a physiological “warning system.” However, in the presence of tissue damage, a complex pathophysiological process results in inflammation and other changes associated with acute pain. Also, the injury response and acute pain impact on all organ systems. As is the case with all clinical pain, there are physical, psychological, and environmental factors that may be involved to varying degrees in individual patient’s experience of acute pain. Recently, it has emerged that, following trauma or surgery, a considerable percentage of patients with acute pain progress to persistent (chronic) pain.
Pain Transduction and Transmission
The nociceptor relays information on thermal, mechanical, and chemical noxious stimuli via slower unmyelinated (C) fibers and small, lightly myelinated (A-delta) fibers to the dorsal horn of the spinal cord at the respective corresponding segmental level or rostrally and caudally passing through Lissauer’s tract. Modulation of this signal at the dorsal horn involves both inhibitory and excitatory mechanisms through local and supraspinal inputs, before transmission to supraspinal structures involved in the perception of pain (Siddall and Cousins 2009). The discovery of transduction mechanisms for heat/capsaicin (TRPV1) and for chemical stimuli (ASIC) has been a major advance that opens up new avenues for peripheral blockade of acute pain (see Siddall and Cousins 2009).
Peripheral Sensitization
Surgical trauma or the noxious stimuli associated with pain are associated with an inflammatory response. A complex range of mediators is released from damaged tissue and inflammatory cells. In addition, nociceptive stimulation activates the neuronal release of substance P, CGRP, and neurokinin A, causing neurogenic inflammation through action on blood vessels and further increased mediator release from inflammatory cells. This “inflammatory soup” induces a phenotypic change within the nociceptor leading to peripheral sensitization (Fig. 7.1), where low-intensity non-noxious stimuli may be perceived as painful. This zone of primary hyperalgesia is also marked by a reduction in thermal threshold detection, which will differentiate it from secondary hyperalgesia (Siddall and Cousins 2009).
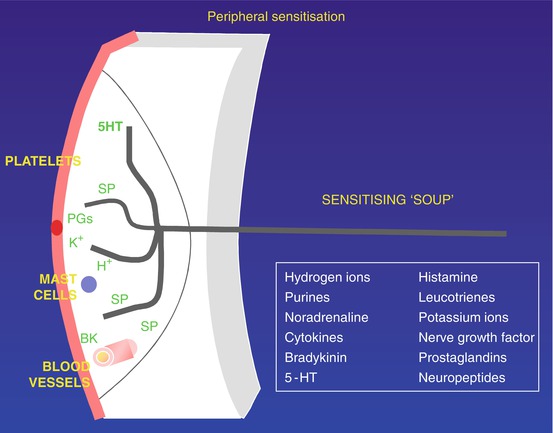
Fig. 7.1
Peripheral sensitization following injury. Injury results in release of potassium (K+) from damaged cells, as well as prostaglandins. K+ strongly activates nociceptive free nerve endings (Аδ- and C-fibers). Prostaglandins sensitize nociceptors to activators such as K+. Antidromic stimuli in collaterals activate release of substance P (SP) (and calcitonin-gene-related peptide [CGRP]; not shown), which plays a pivotal role at three sites: SP (and CGRP) increase capillary permeability, thus allowing the peptide bradykinin to cross capillary walls and strongly activate nociceptors, in turn releasing more SP; SP acts on platelets to release 5-hydroxytryptamine (5HT), which sensitizes nociceptors, again releasing more SP; and SP acts also on mast cells to release histamine, sensitizing nociceptors. The foregoing three SP processes set up vicious circles of increasing sensitization. PGs prostaglandins
Sympathetic Nervous System
The sympathetic nervous system is also implicated in maintenance of acute pain. Inflammation can result in the sensitization of primary nociceptive afferent fibers by prostanoids that are released from sympathetic fibers (Levine et al. 1993). Furthermore, activation of alpha-adrenoreceptors on afferent fibers and innervation of the dorsal root ganglia by sympathetic terminals (McLachlan et al. 1993) can lead to sympathetically mediated pain. Pain syndromes, like complex regional pain syndrome, are associated with features of autonomic dysfunction including vasomotor, sudomotor, and trophic changes (Fig. 7.2) (Binder and Baron 2009).
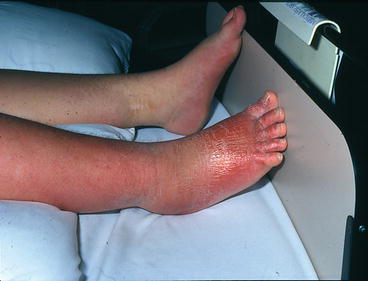
Fig. 7.2
Sympathetic nervous system and acute pain. Patient with CRPS
Central Sensitization
A zone of secondary hyperalgesia with normal thermal threshold in undamaged tissue cannot be explained by peripheral sensitization. Rather, afferent noxious stimuli induce changes in the dorsal horn spinal neurons. This is marked by changes in neuropeptide, neurotrophins, and cytokine levels; receptor activation; and expression, especially the NMDA receptor. Anatomical changes have also been implicated in the pathogenesis of severe central sensitization, with cell death and axonal sprouting. There is also evidence that NMDA receptors are involved in a number of phenomena that may contribute to the medium- or long-term changes that are observed in the transition from acute to chronic pain states. These phenomena include the development of “windup” (Davies and Lodge 1987) facilitation, central sensitization (Woolf and Thompson 1991), changes in peripheral receptive fields, induction of oncogenes, and long-term potentiation (Fig. 7.3) (Collingridge and Singer 1990). Long-term potentiation, in particular, refers to the changes in synaptic efficacy that occur as part of the process of memory and may play a role in the development of a cellular “memory” for pain or enhanced responsiveness to noxious inputs. Furthermore, it appears that NMDA antagonists can attenuate these responses (Woolf and Thompson 1991), indicating a role for NMDA antagonists in the treatment of acute pain and in the prevention of chronic pain states.
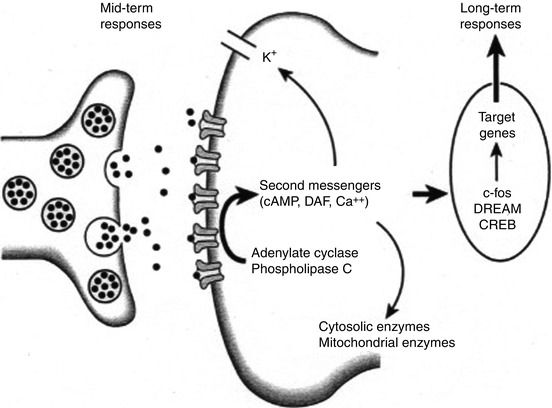
Fig. 7.3
Neurotransmitter release from the central terminal of peripheral afferents results in activation of receptor sites on the postsynaptic membrane. Activation of phospholipase C and adenylate cyclase leads to the production of the second messengers cyclic adenosine monophosphate (cAMP) and diacylglycerol (DAG). Mobilization of these second messengers may result in a decrease in potassium (K+) efflux and elevation of intracellular calcium. The increase in intracellular calcium results in the induction of the proto-oncogene c-Fos, production of Fos protein, and a presumed action on target genes to alter long-term responses of the cell to further stimuli. DREAM downstream regulatory element antagonist modulator, CREB cyclic adenosine monophosphate (cAMP) response element-binding protein
The activation of NMDA receptors appears to set in train a cascade of secondary events in the cell which has been activated. These events lead to changes within the cell, which increase the responsiveness of the nociceptive system and lead to some of the phenomena described above (Dickenson 1996). The NMDA receptor calcium channel in its resting state is “blocked” by a magnesium “plug.” Priming of the NMDA receptor by co-release of glutamate and the peptides acting on the neurokinin receptors leads to removal of the magnesium plug and subsequent calcium influx into the cell leading to secondary events such as oncogene induction (Morgan and Curran 1986), production of nitric oxide (NO), (Garthwaite et al. 1988), and activation or production of a number of second messengers including phospholipases, polyphosphoinosites (IP3, DAG), cyclic guanine monophosphate (cGMP), eicosanoids, and protein kinase C. These second messengers then act directly to change the excitability of the cell or induce the production of oncogenes, which may result in long-term alterations in the responsivity of the cell. Prolonged stimulation, presumably through sustained and therefore excitotoxic release of glutamate, may result in cell death (Fig. 7.4).
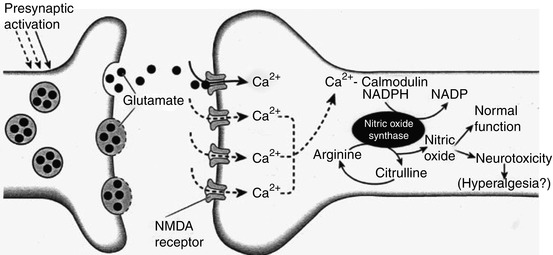
Fig. 7.4
Diagram illustrating postsynaptic events following release of glutamate from central terminals of primary afferents in the spinal cord. Following priming of the N-methyl-D-aspartate (NMDA) receptor complex, subsequent glutamate release results in NMDA receptor activation with subsequent calcium influx. Intracellular calcium then acts on the calmodulin-sensitive site to activate the enzyme nitric oxide synthase (NOS). In the presence of a cofactor nicotinamide adenine dinucleotide phosphate (NADPH), NOS uses arginine as a substrate to produce nitric oxide and citrulline. Nitric oxide has a role in normal cellular function, but increased production may be involved in hyperalgesia and may lead to neurotoxicity
The exact role of NO in nociceptive processing is still unclear (Malmberg and Yaksh 1993). However, the production of NO is implicated in the induction and maintenance of chronic pain states (Meller and Gebhart 1993) and may be one of the factors responsible for the cell death and reduced GABA inhibition which has been demonstrated to occur under these conditions. It has been suggested that NO acts as a positive feedback mechanism in the maintenance of pain. The changes in nociceptive processing, gene expression, and others to be described below form a continuum with the early effects of nociception. Thus, it is no longer logical to view acute, subacute, and chronic pain as being entirely separate.
The production of arachidonic acid metabolites as part of the cascade that occurs following NMDA receptor activation also raises an interesting potential avenue of intervention that is already being explored (Eisenach 1993). Although the peripheral effects of NSAIDs have been emphasized in the past, it appears that there may be a role for the spinal administration of NSAIDs. Spinal NSAIDs act either directly on receptors, such as the strychnine-insensitive glycine site of the NMDA receptor complex, or influence the production of metabolites within the cell.
Descending Inhibition
The foregoing processes mostly result in enhanced peripheral and spinal cord nociception with the potential to progress to persistent pain. Fortunately, a powerful descending modulatory process “descending inhibition” provides potent inhibitory input at various levels, including the dorsal horn, via release of gamma-aminobutyric acid (GABA) and other inhibitory neurotransmitters.
Visceral Pain
Visceral pain differs in many distinct ways to somatic pain. For instance, visceral pain does not always indicate tissue damage, is poorly localized, can show a referred component, and can be associated with autonomic and affective components. Distension and spasm of visceral hollow organs can produce pain. Nonspecific visceral receptors that use an intensity coding mechanism convey afferent impulses via a limited number of autonomic afferent fibers to the dorsal horn, where they branch, forming synapses with dorsal horn neurons at different segments. The visceral structures represent a large percentage of the body surface area and however represent only 10 % of the fiber types in the dorsal horn. Therefore the somatotopic map of the viscera is poor leading to indistinct localization of pain. The convergence of visceral afferents onto dorsal horn neurons (viscero-somatic convergence) supplied by cutaneous and deep somatic afferents gives rise to the characteristic referral pattern of visceral pain. Similarly, augmentation of pain due to sensory interaction between different visceral structures that share afferent circuitry (viscero–viscero convergence) can occur.
The Pain Response
Injury Response
The pathophysiology of the injury response is wide-ranging, resulting in impairment of homeostasis, brought about by the neuroendocrine release and local cytokines. A hypercatabolic state ensues, with mobilization of glucose stores, protein and fat breakdown, and electrolyte and water flux (Carli and Schricker 2009).
Neuroendocrine and Metabolic Response
Catabolic
Increase in ACTH, cortisol, ADH, GH, catecholamines, renin, angiotensin II, aldosterone, glucagon, and interleukin-1
Anabolic
Due to decrease in insulin and testosterone
Metabolic
Carbohydrate
Decrease in insulin secretion/action
Hepatic glycogenolysis (epinephrine, glucagon)
Gluconeogenesis (cortisol, glucagon, growth hormone, epinephrine, free fatty acids)
Hyperglycemia, glucose intolerance, and insulin resistance
Protein
Muscle protein catabolism and increased synthesis of acute-phase proteins
Due to increase in cortisol, epinephrine, glucagon, and interleukin-1
Fat
Increased lipolysis and oxidation
Due to increase in catecholamines, cortisol, glucagon, and growth hormone
Water and Electrolyte Flux
Retention of H2O and Na+, increased excretion of K+, and decreased functional extracellular fluid with shifts to intracellular compartments (data from Kehlet (1989))
Cardiovascular Response
Sympathetic stimulation results in increased cardiac output and peripheral vascular resistance. In the compromised heart, this increased cardiac workload together with the reduction in myocardial oxygen supply through the decreased diastolic filling time will be deleterious (Wu and Liu 2009). The relationship between postoperative pain and noxious stimuli has been illustrated by the effect of epidural analgesia in preventing and reversing these cardiovascular abnormalities (Sjogren and Wright 1972; Hoar and Hickey 1976; Kumar and Hibbert 1984; Wu and Liu 2009). There is impressive evidence from animal studies that noxious stimulation results in coronary artery vasoconstriction and potential for myocardial ischemia. This has been supported by evidence in recent systematic reviews with reduction in cardiac morbidity associated with epidural analgesia, particularly in intra-abdominal aortic surgery (Wu and Liu 2009).
Respiratory Response
Respiratory dysfunction results through a variety of mechanisms. Noxious stimuli can evoke both involuntary spinal reflexes with muscle spasm and voluntary reduction in muscle movement due to perception of pain. This muscle splinting impacts on ventilatory parameters, leading to small airway closure, and impairment of cough, leading to atelectasis and ensuing hypoxia (Wu and Liu 2009). Substantial correction of the majority of the abnormalities in pulmonary function can be obtained with effective pain relief associated with epidural block (Bowler et al. 1986; Scott and Kehlet 1988). The cumulative evidence of many clinical trials, when subjected to meta-analysis, has confirmed the superiority of epidural over systemic opioid administration in minimizing postoperative pulmonary morbidity.
Gastrointestinal Effects
Increased sympathetic activity results in decreased bowel motility and increased intestinal secretions, although this can also be attributed to opioid effects. There is some evidence that pain relief with neural blockade may reduce the transit time of x-ray contrast media through the gut, from up to 150 h in a control group to 35 h in a group receiving epidural analgesia (Ahn et al. 1988). There is also evidence that the pain-related impairment of intestinal motility may be relieved by epidural local anesthetic, but not by epidural opioid (Scheinin et al. 1987; Thoren et al. 1989, 1992; Wattwil et al. 1989; Liu et al. 1995a, b; Wu and Liu 2009). Interestingly, there is good evidence that the systemic, intravenous, administration of lignocaine speeds the return of bowel function after radical prostatectomy, as well as reducing pain and shortening hospital stay (Groudine et al. 1998).
Immunological Effects
Immunological effects with suppression of humoral and cellular immunity and coagulation abnormalities have also been recognized (Wu and Liu 2009). Changes in coagulation and fibrinolysis associated with major surgery may be partly modified by pain relief with neural blockade (Jorgensen et al. 1991). However, interpretation of these results is complex, because factors other than pain may be involved. Also, the absorption of local anesthetics associated with neural blockade may result in an antithrombotic effect (Kehlet 1998). Encouragingly, a clinical study comparing the effects of epidural and general anesthesia in peripheral vascular surgery has found that the epidural group needed fivefold fewer repeat operations for graft failure within 1 month (Christopherson et al. 1993). Changes in immunocompetence and acute-phase proteins are well documented in association with surgical trauma. Pain relief with neural blockade has a mild influence on various aspects of the surgically induced impairment of immunocompetence. The mechanism has not been completely elucidated, but may be partly explained by the concomitant inhibition of various endocrine metabolic responses. It is currently not clear if the mechanism of this effect is predominantly a result of blockade of nociception. Elucidation of this mechanism is important because posttraumatic immunodepression has been impossible to modulate by other therapeutic measures (Kehlet 1998; Wu and Liu 2009).
Attenuation of the Injury Response
Adequate management of postoperative pain is essential to attenuate the injury response. The role of different factors in the modification of the injury response is reviewed by Kehlet (1996, 1998), Liu et al. (1995b), and Wu and Liu (2009). While favorable data exist for effects on outcome of neural blockade using local anesthetic and opioid for lower-abdominal and lower-extremity procedures, less convincing effects are seen for upper abdominal and thoracic procedures.
Psychological Factors Affecting the Acute Pain Response
There is now considerable evidence that psychological factors account for a significant proportion of the variance in pain response. In addition, unrelieved pain leads to psychological changes, with increased anxiety, sleep disturbances, low morale, loss of control, and inability to think and interact (Cousins et al. 2004; Katz and Melzack 2009). Anxiety is the psychological variable which is most reliably related to high levels of pain. Circumstances associated with acute postoperative pain are probably some of the most potent in aggravating such fears. Fear of the unknown is also a major component of the general anxiety which patients experience. Hospitalization itself produces many threats, including possible disability, loss of life, coping with a new situation, loss of normal freedom, and separation from one’s family and normal routine (Johnson 1980; Katz and Melzack 2009).
Preoperative anxiety has been shown to be associated with higher levels of pain in the early postoperative period after abdominal (Caumo et al. 2002), coronary artery bypass surgery (Nelson et al. 1998), and 1 year after total knee replacement (Bradner et al. 2002).
Higher preoperative pain catastrophizing scores correlated with higher pain scores immediately postoperatively after anterior cruciate repair and breast surgery (Pavlin et al. 2005; Jacobsen and Butler 1996).
Higher preoperative pain catastrophizing scores correlated with higher analgesic use after breast surgery (Jacobsen and Butler 1996).
The effects of situational or environmental variables have been shown to be important, as exemplified by the powerful effect of anxiety and perceived control over the situation (Peck 1986). Some patients may interpret the use of sophisticated monitoring equipment as implying that their situation is one of the imminent disasters (Cousins 1970). The anxiety experienced by family members is often transferred directly to the patient and serves to reinforce or reactivate his or her own fears. Another important area of investigation has been the interaction between individual and situational variables, such as the interaction between coping style and the control the patient has over the situation (Andrew 1970).
Placebo and expectation effects can sometimes play a very powerful role. One aspect of these effects is the patient’s confidence and belief that the healthcare professional will be able to provide pain relief, and clearly such a placebo response is augmented by a positive doctor–patient or nurse–patient relationship (Dimatteo and Di Nicola 1982).
Studies suggest that the initial relief experienced in a new situation may be an important determinant of future relief, because the patient’s expectations may be conditioned at that time. On the other hand, inadequate relief may condition a negative expectation, which could adversely affect later pain control. This indicates the importance of providing adequate pain control as quickly as possible and conveying the expectation that the pain control procedures will continue to provide effective pain relief (Voudoris and Peck 1985; Katz and Melzack 2009).
Acute Postoperative Neuropathic Pain
Neuropathic pain is pain initiated or caused by a primary lesion (or dysfunction) in the nervous system (Merskey and Bogduk 1994). In the postsurgery or posttrauma patient, neuropathic pain may have an early onset within the first 24 h; this is contrary to classic teaching. There may also be a delayed onset of the order of 10 days. Numerous physiological and structural changes occur following nerve injuries and have been summarized in several review articles (Siddal and Cousins 2008; Woolf and Manion 1999). Experimental evidence concerning the mediators involved in nerve injury and central sensitization is numerous. Injured sensory neurons undergo Wallerian degeneration with myelin sheath disruption, invasion by immune cells, and distal degeneration of the axon before regeneration can occur. Many changes have been implicated including neuroma formation, increased or novel expression of sodium channel subtypes leading to ectopic pacemaker-like activity, upregulation of voltage-gated channel expression, and neuro-immune interactions both peripherally and centrally. These afferent barrages, alterations in inhibitory and anatomical changes, lead to marked changes in the dorsal horn neurons that may lead to persistent central sensitization. Recognition of neuropathic pain and early aggressive treatment are therefore paramount to prevent long-term problems. Features suggestive of neuropathic pain are:
(Adapted from Acute Pain Management: scientific evidence NHMRC 1999)
Pain in the absence of ongoing tissue damage.
Pain in an area of sensory loss.
Paroxysmal or spontaneous pain.
Allodynia (pain in response to non-painful stimuli), e.g., “tingling” and “pins and needles.”
Hyperalgesia (increased pain in response to painful stimuli).
Dysesthesias (unpleasant abnormal sensations).
Characteristic of pain different from nociception: burning, pulsing, stabbing pain
Sometimes a delay in onset of pain after nerve injury (note: some neuropathic pain has immediate onset).
Hyperpathia: increasing pain with repetitive stimulation; “after response” (continued exacerbation of pain after stimulation); and radiation of pain to adjacent areas after stimulation.
Tapping of neuromata (spontaneously firing growth buds from damaged peripheral nerves) produces a radiating electric shock sensation in the distribution of the nerve (Tinel’s sign).
Poor response (not unresponsiveness) to opioids.
Presence of a major neurological deficit (e.g., brachial plexus avulsion, spinal injury).
Despite these features neuropathic pain remains a significant diagnostic challenge. There is no single diagnostic test for neuropathic pain. Nerve conduction velocity tests and electromyography provide information about large myelinated fibers, but not about the small nociceptive afferents. Quantitative sensory testing is a psychophysical test that informs about large and small fiber function through subjective appreciation of detection thresholds to touch, pain, thermal, and vibration stimuli. Its use is limited in daily clinical practice due to the specialized equipment and training needed. Many neuropathic assessment tools have been validated for the discrimination of neuropathic pain. The S-LANSS (Leeds Assessment of Neuropathic Symptoms and Signs pain scale) score is a simple and valid seven-item self-reporting tool with sensitivity of 74 % and specificity of 76 % (Table 7.1). While these tools can aid in diagnosis, the “gold standard” at present is clinical history and examination.
Table 7.1
Neuropathic pain screening tools
Pricking, tingling
Stay updated, free articles. Join our Telegram channel
Full access? Get Clinical Tree
 Get Clinical Tree app for offline access
Get Clinical Tree app for offline access

|