CHAPTER 18 Acute myeloid leukemias
Introduction
The acute myeloid leukemias (AML) are malignancies of immature precursors of the non-lymphoid hematopoietic lineages, involving the bone marrow (BM), peripheral blood (PB), and possibly other tissues. Taken together, this heterogeneous group of neoplasms has an estimated incidence between 1.6 and 3.7 per 100 000 population per year in the US and Western Europe.1–3 Yearly incidence rates appear to increase with patient age, and the median age at diagnosis is 65.2 AML is the predominant form of acute leukemia in adults, but accounts for only 15–20% of acute leukemias in pediatric patients.4 Environmental exposures or therapies that damage DNA, and a number of clinical syndromes affecting DNA repair pathways or tumor suppressor genes lead to increased risk of AML. Intensive study has revealed many somatic genetic lesions that appear to contribute to the development of AML. Approximately 20% of cases show recurrent cytogenetic abnormalities, while fewer than 10% of cases have a normal karyotype and fail to show mutations in genes such as FMS-like tyrosine kinase 3 (FLT3), nucleophosmin (NPM1), CCAAT/enhancer-binding protein alpha (CEBPA) and myeloid/lymphoid or mixed lineage leukemia (MLL) that are commonly altered in AML.5,6 High-resolution studies of genomic copy number alterations in AML with comparison to normal somatic cell DNA from the affected patients, as well as initial full-genome sequencing efforts have given indications that there may be additional recurrent genetic lesions in this malignancy, as well as many other mutations that may be very rare, pathogenic only in concert with numerous other mutations, or entirely unrelated to pathogenesis.7–9 Prognosis of patients with AML shows a striking relationship with the cytogenetic and other mutational features of the malignant blasts, with the implication that a complete diagnostic report for AML should contain this information, despite the logistical challenges of combining data from several laboratory disciplines into a unified synopsis.
Classification systems
The rapid evolution of classification schemes for AML has resulted from several successive waves of new technology entering the clinical hematology laboratory and being validated in clinical trials. The French-American-British (FAB) classification of 1976 and its later modifications primarily relied upon morphologic examination, cytochemical tests, and limited immunophenotyping to establish lineage-related classification of myeloid blasts. In the years that followed, additional data including recognition of common cytogenetic abnormalities, clinical history and correlation with clinical outcomes led to the World Health Organization (WHO) classification scheme in 2001, with distinct AML categories defined by: 1) recurrent cytogenetic abnormalities; 2) presence of myelodysplastic changes; 3) history of methylating agent or topoisomerase 2 inhibitor chemotherapy drug exposure, or 4) a ‘not otherwise specified’ category encapsulating the FAB morphologic/cytochemical classification.10 The defining BM blast percentage for AML was decreased from 30% to 20% at this time, largely because myelodysplastic syndrome (MDS) patients with over 20% blasts had been found to have a poor prognosis very similar to those with AML.11 The transition from FAB to WHO 2001 was a pragmatic effort to generate a clinically useful classification system providing prognostic information relevant to the therapeutic options available, while incorporating cytogenetic findings likely related to disease pathogenesis, and still retaining familiar entities defined by their morphology and antigen expression. Incorporating the response to therapy as part of the basis for the classification scheme may necessitate future revisions if new therapies with distinctive properties come into wide use. For the practicing hematopathologist, knowledge of the preceding classification systems is essential, given that long-running clinical trials often refer to the older systems, and reports with the correct current classification and parallel classification by the older terminology are appreciated by clinicians and trial researchers.
A revised WHO classification was published in 2008 (Table 18.1), and builds on the insights of the 2001 system with an increased number of recurrent cytogenetic abnormalities, and the creation of provisional categories for AML cases with mutations in the NPM1 or CEBPA genes.6 The key role of cytogenetic data is further increased by the stipulation that cases with the t(8;21)(q22;q22), inv(16)(p13.1q22), t(16;16)(p13.1;q22), or t(15;17)(q22;q12) need not have >20% blasts to be diagnosed as AML. Additional weight is given to cytogenetic abnormalities in the AML with myelodysplasia-related changes category, where specific MDS-associated cytogenetic findings can take the place of morphologic evidence of myelodysplasia. Two entities found in patients with Down syndrome are now classified separately, and the therapy-related AML category has been simplified to a single entity for patients with history of exposure to any of a broad range of cytotoxic drugs or ionizing radiation therapy. Adjustments to the criteria for AML with myelodysplasia-related changes, and the addition of blastic plasmacytoid dendritic cell neoplasm as an AML-related precursor neoplasm are the other significant changes from the WHO 2001 scheme.6 In the sections that follow, we will use the WHO 2008 framework to organize discussion of these malignancies, after a brief review of the clinical features and laboratory methods required for their analysis.
Table 18.1 World Health Organization classification of acute myeloid leukemia (AML) and related precursor neoplasms
Methods for diagnosis and classification
Peripheral blood and bone marrow cell count and morphology
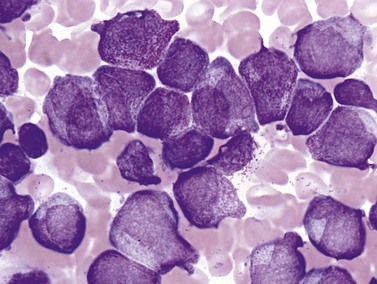
Complete blood counts and morphologic review of PB smears usually reveal multiple abnormalities in AML patients. Reduced counts of red cells, platelets and the normal leukocytes are commonly seen, but the white blood cell count (WBC) is most often elevated due to circulating blasts. Patients usually present before the WBC count rises above 50 × 109 cells per liter, but a minority of patients have WBC counts over 100 × 109 cells per liter, and occasional cases show WBC counts as low as 1 × 109 cells per liter with few circulating blasts. BM evaluation should be performed from aspirate smears of adequate quality reviewed in conjunction with bone marrow trephine biopsy (BMTB) sections. With the exception of a few cytogenetically-defined subcategories, blasts in the PB or BM must be ≥20% of total nucleated cells for a diagnosis of AML, based on a 200-cell count in the blood or a 500-cell count in the BM. The presence of Auer rods (linear filaments of primary granules) (Fig. 18.1) is the only definitive morphologic finding sufficient to distinguish myeloid blasts from lymphoid ones, but other features typical of myeloid blasts include larger blast size, fine chromatin, multiple often easily visualized nucleoli and cytoplasmic granulation. Blasts of some cases of AML (corresponding to those formerly classified in the FAB M0 and M1 categories) may be morphologically similar to lymphoblasts, small to medium in size with few nucleoli and minimal amounts of agranular cytoplasm, while others (corresponding to monoblasts and promonocytes in the FAB M4 and M5 categories) show monocytic differentiation, being large in size with round to delicately folded or convoluted nuclei, fine lacy chromatin, visible nucleoli and basophilic cytoplasm with absent to minimal granulation. A background of dysplasia in other BM myeloid lineages may also provide evidence that blasts are more likely to be of myeloid type. In some subcategories of AML, modifications of the blast count are part of the WHO 2008 classification: promonocytes are considered as blasts in AML cases with myelomonocytic, monoblastic or monocytic differentiation; the abnormal promyelocytes in APL are considered blasts; and in AML not otherwise specified erythroid/myeloid erythroleukemia cases, the blast percentage is calculated from non-erythroid BM nucleated cells.6 Patients with myeloid sarcomas are considered to have AML regardless of PB or BM blast counts.
Immunophenotyping
Flow cytometry (FCM) is the preferred method of characterizing surface and internal antigen expression in leukemia cases, as modern flow cytometers and fluorescent antibodies enable four or more antigens to be measured simultaneously on each cell assayed, together with forward and side light scatter properties that correlate with cell size and internal complexity or granularity. Given the large number of different antigens that may be expressed by AML or other leukemic blasts, panels of antibodies used for diagnosis can be extensive, and are usually designed with one common antibody (typically binding the leukocyte marker CD45) present in all tubes used for staining cells, so that cell populations defined by CD45 expression and forward or side light scatter can be uniformly compared across all antibody stains in the panel. Myeloid surface antigens include CD13, CD33, CD117 (for more immature precursors), and CD15 (for more mature cells), while monocytic markers include CD14 and CD64. Megakaryocytic lineage is suggested by the platelet antigens CD41a and CD61, while erythroid markers include CD71, glycophorin and hemoglobin A. Surface expression of CD34 is a broad marker of immaturity seen in myeloid and lymphoid lineage blasts. Internal antigens helpful for characterizing myeloid blasts include myeloperoxidase (MPO) and TdT, which, as its role in lymphoid biology would suggest, is usually negative in AML but can be expressed aberrantly in a subset of cases. Many AML cases can be demonstrated to express antigens of other hematopoietic lineages: for example, t(8;21)-containing cases often show expression of the B cell antigens CD19, Pax5 or cytoplasmic CD79a; and T cell markers CD2 or CD7 are not uncommon in several subtypes of AML.12 The aberrant expression of such markers typically does not warrant a diagnosis of biphenotypic or mixed phenotype acute leukemia (see Chapter 19), but may increase the sensitivity and specificity of detecting residual or recurrent disease following therapy.
Cytochemistry
A key part of lineage determination in the FAB classification scheme, cytochemical testing on air-dried blood or BM aspirate smear slides is performed to a more limited degree in most modern laboratories, with the result that many leukemias may not be fully evaluated by FAB criteria under current protocols.13 Cytochemical stains for myeloperoxidase (MPO) and the somewhat less specific Sudan Black B (SBB) are used to identify myeloid lineage, although these stains are negative in very early myeloblasts and monoblasts, as well as in erythroid and megakaryocytic blasts. SBB has the advantage of being usable even when the air-dried smears have not been recently prepared. The nonspecific esterase (NSE) activity that characterizes monocytes and monoblasts can be detected by reactivity with alpha naphthyl butyrate esterase, or sodium fluoride-inhibited reactivity with alpha naphthyl acetate esterase. Naphthol-ASD-chloroacetate esterase (CAE) activity is specific for neutrophil and mast cell lineages. Using these stains, AML with minimal differentiation cases are defined by having less than 3% of blasts positive for MPO, SBB or CAE, and lacking NSE activity; the designation of these cases as acute myeloid leukemias relies on immunophenotypic detection of expression of some combination of CD13, CD117 or CD33 and lack of lymphoid differentiation markers. AML without maturation is distinguished from AML with minimal differentiation by having >3% MPO or SBB-positive blasts, or the presence or Auer rods (which should also stain for both MPO and SBB). Monoblasts and promonocytes seen in the acute myelomonocytic or acute monoblastic or monocytic leukemia categories show blast-equivalent cells with NSE reactivity but usually no MPO or SBB reactivity. Acute megakaryoblastic leukemias may show NaF-resistant NSE activity for alpha naphthyl acetate, but are MPO and SBB negative.
Cytogenetics and molecular testing
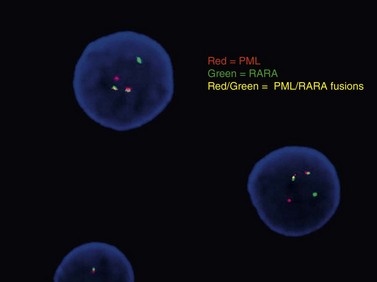
Conventional karyotype analysis, fluorescence in situ hybridization (FISH) assays for detecting translocations, and molecular biology tests for point mutations or other small genetic lesions have become an integral part of evaluating and sub-classifying acute myeloid leukemias, and can guide therapy, most notably for APL carrying t(15;17) that respond to all-trans retinoic acid (ATRA) treatment (Fig. 18.2). Cytogenetic and molecular testing additionally impacts therapeutic decision-making for patients with favorable cytogenetic or gene mutation-containing leukemias such as cases with low WBC counts and t(8;21), inv(16), t(16;16), or NPM1 mutation without cytogenetic abnormalities and without FLT3-internal tandem duplication, in which allogeneic stem cell transplant does not appear to confer increased survival.14,15 The key cytogenetic and mutational lesions in AML will be discussed in greater detail in the following sections dealing with each classification category.
Acute myeloid leukemia with recurrent genetic abnormalities
These leukemias feature balanced translocations or inversions, or belong to new provisional categories defined by mutation of the NPM1 or CEPBA genes. These cytogenetic or mutationally-defined features have been associated with prognostic significance in clinical trials (Table 18.2). Most cases with recurrent translocations or inversions have some characteristic morphologic, immunophenotypic or clinical features.16–19 If the classic t(8;21), inv(16), t(16;16), or t(15;17) translocations are present, 20% blood or BM blasts are not required for diagnosis. It should be noted that AML in patients with a history of chemotherapy or radiation therapy should be classified in the therapy-related AML category regardless of other findings. Multilineage dysplasia is commonly seen in the recurrent translocation/inversion cases with inv(3), t(3;3), or t(6;9), but the cytogenetic findings take priority for classification of these entities. Since the latter chromosomal changes were found also in patients clinically presenting as MDS, cases with inv(3), t(3;3), or t(6;9) can be classified as AML only when ≥20% blasts are present.
Table 18.2 AML with recurrent genetic abnormalities: prognostic correlations
| AML with t(8;21)(q22;q22); (RUNX1-RUNX1T1) | Good prognosis |
| AML with inv(16)(p13.1q22) or t(16;16)(p13.1;q22); (CBFB-MYH11) | Good prognosis |
| Acute promyelocytic leukemia (APL) with t(15;17)(q22;q12); (PML-RARA) | Good prognosis |
| AML with t(9;11)(p22;q23); (MLLT3-MLL) | Intermediate prognosis; better than AML with other 11q23 translocations |
| AML with t(6;9)(p23;q34); (DEK-NUP214) | Poor prognosis, similar to AML with myelodysplasia-related changes |
| AML with inv(3)(q21q26.2) or t(3;3)(q21;q26.1); (RPN1-EVI1) | Poor prognosis |
| AML (megakaryoblastic) with t(1;22)(p13;q13); (RBM15-MKL1) | Probably good prognosis with intensive AML chemotherapy |
| AML with mutated NPM1 (provisional) | Good prognosis |
| AML with mutated CEBPA (provisional) | Good prognosis |
AML with t(8;21)(q22;q22); (RUNX1-RUNX1T1)
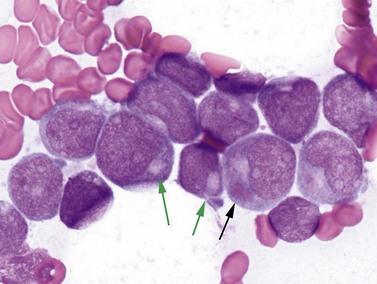
Cases with the t(8;21) (q22;q22) translocation most commonly present in younger patients, and comprise 5–10% of AML diagnoses. Morphologic features are typically consistent with the former FAB M2 category (‘with maturation’) with blasts displaying ample cytoplasm with abundant granules and some blasts containing Auer rods (Fig. 18.3). Telltale features suggesting this diagnosis include prominent perinuclear hofs, the presence of larger pink granules among the more typical darker cytoplasmic granules, and frequent aberrant expression of B-cell antigens such as CD19, Pax5, or cytoplasmic CD79a. Other morphologic findings that can be seen are Auer rods in more mature granulocytes, abnormal very large granules in blasts, and varying degrees of dyspoietic morphology in granulocytes, including Pelger–Huetoid bilobed neutrophils. CD34 (often overexpressed) and myeloid antigens CD13, CD33 and MPO are usually positive by FCM, although CD33 may be weak. The RUNX1 gene (synonyms: AML1, CBFA) encodes the core binding factor alpha protein (CBFA), one component of a heterodimeric hematopoietic transcription factor; the abnormal fusion to the RUNX1T1 gene (synonym: ETO, for eight-twenty-one) likely impairs or misdirects gene-regulatory activity.20 Prognosis for these cases is good with modern therapies, although high presenting WBC counts (>20 × 109 cells per liter) and KIT mutations (see below) lead to an intermediate prognosis.
AML with inv(16)(p13.1q22) or t(16;16)(p13.1;q22); (CBFB-MYH11)
The inv(16) or t(16;16) cases are also predominantly diagnosed in younger patients and account for 5–10% of all AML. Lesions affecting 16p13.1 involve the gene for the second half of the core binding factor (CBF) transcription factor (CBFB core-binding factor beta) fused to a smooth muscle myosin heavy chain gene (MYH11), possibly indicating an underlying biological basis for the good prognosis and younger demographic that these cases share with t(8;21) AML.21 Clinically, these leukemias show a high rate of extramedullary disease, approaching 50% of cases. The morphologic features of inv(16) or t(16;16) AML are highly distinctive, with most but not all cases showing granulocytic or monocytic blasts (often consistent with FAB M4 criteria) accompanied by abnormal eosinophil precursors in the BM (Fig. 18.4). The eosinophil precursors at the promyelocyte and myelocyte-equivalent stages show occasional to numerous large purple or basophilic granules of variable shape, intermixed with eosinophilic granules. FCM often documents several blast populations with myeloid or monocytic marker expression, and frequent (but nonspecific) aberrant expression of the T-cell marker CD2. FISH or real-time PCR (RT-PCR) testing is more sensitive than conventional karyotyping for detection of the inv(16)(p13.1q22) abnormality.
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


