CHAPTER 19 Acute lymphoblastic leukemia/lymphoma and mixed phenotype acute leukemias
Introduction
Acute leukemias are clonal malignant diseases of early hematopoietic progenitor cells. The lymphoblastic forms (acute lyphoblastic leukemia, ALL) are characterized by homogeneous blast cell populations. The accurate diagnosis and classification of acute lymphoblastic and mixed phenotype acute leukemias requires multiple diagnostic techniques1 including:
Acute lymphoblastic leukemia/lymphoma
Clinical presentation
Symptoms may be of abrupt onset, or more insidiously, occurring over a period of weeks or even months. Patients usually present clinical signs of BM failure. Tiredness and pallor are induced by anemia. Infections are linked to the underlying neutropenia. Purpura, bruising or gum bleeding are related to thrombocytopenia. Fever may be present, and half of affected children have bony pain. Differential diagnosis will include infections, immune diseases, macrophage disorders, congenital BM diseases, aplastic anemia and secondary infiltration of other tumors.2 Less commonly, lymphadenopathy, splenic or liver involvement, the symptoms associated with a mediastinal mass or CNS disease may be the presenting features of ALL. Rarely, testicular or other tumor-forming presentation may be the first sign of the disease.
Most patients present with anemia and/or thrombocytopenia. Approximately 25% of patients have a leukocyte count of <5 × 109/l, 50% between 5 and 50 × 109/l, and 25% >50 × 109/l. Leukocyte counts >100 × 109/l may lead to leukostasis and should be treated as a medical emergency.3 High blast counts are more commonly found in T-ALL patients.
At the time of diagnosis the BM is usually hypercellular, with replacement of normal hematopoiesis by the blast cells. Rare patients present marrow hypoplasia, before developing a typical leukemic picture.4 Such patients may improve spontaneously or respond transiently to aplasia-directed immunosuppressive treatment before leukemia becomes apparent.
The term lymphoblastic lymphoma (LBL) is used when the disease is confined to an extramedullary infiltrate with no or minimal involvement of PB and BM. Patients with T-LBL constitute above 90% of LBL cases and usually present with signs associated with a mediastinal mass.5 Patients with B-LBL often present skin or head and neck lesions and are usually asymptomatic.
Classification
The French-American-British (FAB) classification of acute lymphoblastic leukemia introduced in 1976 was based on morphology alone and recognized three types of blast cells in ALL, namely: small homogeneous blasts with round nuclei and scanty cytoplasm (L1), larger blasts with irregular nuclei, prominent nucleoli and more abundant cytoplasm (L2) and basophilic cells with prominent cytoplasmic vacuoles (L3).6 The L3 type included most cases previously termed B-ALL and expressing surface-immunoglobulin (SIg). However, some other types of ALL, particularly those with t(1;19), could also present with this cytology. The morphological FAB classification did not have any clinical significance.
The WHO classification in 2001 largely adopted the 1994 revised European–American classification of lymphoid neoplasms (REAL).7 Thus, the WHO classification recognized precursor T- and B-cell neoplasms corresponding to ALL, but excluded cases with the t(8;14)(q24;q32), (and the rarer variants with t(2;8)(p12;q24) and t(8;22)(q24;q11)), which have undergone immunoglobulin gene rearrangement and express surface immunoglobulins. These cases correspond to leukemic presentation of Burkitt’s lymphoma, which in the WHO system was classified with mature B-cell neoplasms. Such cases, if present with peripheral blood involvement, were termed Burkitt cell leukemia.
In the WHO 2008 classification, B-cell lymphoblastic leukemia/lymphomas are defined as neoplasms of precursor cells committed to B-lineage.1 They include B-ALL and B-LBL and are further subdivided by the presence or absence of specific recurring cytogenetic abnormalities (Table 19.1). The following cytogenetic entities have been recognized in B-lymphoblastic leukemia/lymphoma: t(9;22)(q34;q11), t(1;19)(q23;p13), t(12;21)(p12;q22), t(various;11q23 rearranged), hyperdiploid ALL, hypodiploid ALL and B-lymphoblastic leukemia/lymphoma not otherwise specified (NOS). T-lymphoblastic leukemia/lymphomas are defined as neoplasms of precursor cells committed to the T-lineage and include T-ALL and T-LBL.
Table 19.1 WHO 2008 classification of precursor lymphoid neoplasms
Cytology, cytochemistry and histopathology
The morphology of lymphoid blasts should be evaluated on well-prepared May–Grünewald–Giemsa or Romanowsky-stained smears of PB or BM. Cytological appearance is variable (Fig. 19.1). In most cases, the blasts are small to medium-sized with round nuclei and fine but densely packed homogeneous chromatin. Nucleoli are small and usually single or absent. In some patients, blasts can have rather condensed chromatin making distinction from chronic lymphocytic leukemia difficult. The cytoplasm is scanty and weakly basophilic. Cytoplasmic vacuoles may be present. In 10–15% of cases, blasts are larger, with irregular nuclei showing clefting or indentation. Nucleoli are more prominent, often large and occasionally multiple. The cytoplasm is relatively abundant and may be finely reticulated. Basophilia is variable. Scanty azurophil granules are rarely present and usually readily distinguished from granules in myeloid precursors but sometimes similar to those of large granular lymphocytes. A ‘hand-mirror’ variant has been described but is probably of no clinical significance.8
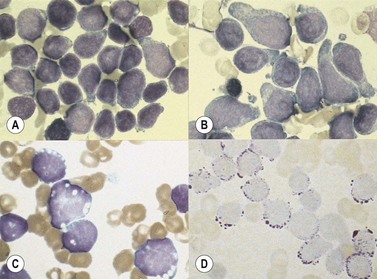
There are no discriminant diagnostic cytochemical stains that distinguish ALL. Lymphoblasts are negative when stained for myeloperoxidase. Very rarely, coarse granular or globular positivity with Sudan Black B may be present. T-lineage ALL frequently shows localized or polar staining of acid phosphatase, but these appearances are not specific. The periodic acid-Schiff (PAS) reaction is useful in identifying lymphoblasts, which usually show fine granules or blocks of positivity (Fig. 19.1). These are found in up to 95% of cases, although they may be very rare, occurring in less than 1% of blasts. The distinctive feature of positivity in lymphoblasts is the absence of any diffuse PAS cytoplasmic staining, which is present in myeloid lineage cells. Lymphoblasts show a clear-glass cytoplasm in which positive granules and blocks are sharply defined. Neutrophils stain strongly and serve as an internal control for the quality of the PAS stain.
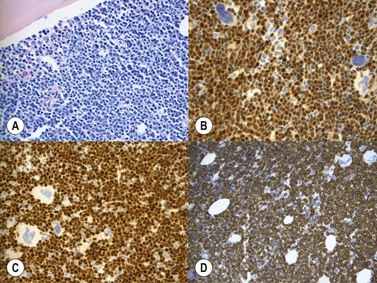
Marrow trephine biopsies usually show maximal cellularity due to the diffuse invasion of sinuses by blast cells (Fig. 19.2A). The lymphoblastic population is usually homogeneous. In the majority of cases, the cells are small with scanty cytoplasm. Nuclei show finely stippled chromatin, often with peripheral condensation. Nucleoli are small and seldom multiple. A minority of cases show larger cells with irregular nuclei and increased cytoplasm. T-lineage ALL may show some degree of nuclear irregularity, and the nuclei may be small and hyperchromatic. Varying degrees of reticulin fibrosis (reversible on successful treatment) may be present in more than half the cases, which can lead to ‘dry-tap’ aspirations of BM. This is more common in B-lineage compared to T-lineage ALL and has been shown to be of poor prognostic significance.9 Areas of marrow necrosis, usually patchy, but occasionally extensive, may be present, and can delay diagnosis if no circulating blasts are present. Cases presenting as aplastic anemia (Fig. 19.3A, B) may show a degree of reticulin fibrosis not generally seen in true aplasia, and are usually of common-B-lineage ALL phenotype. If no material has been submitted for flow cytometry, immunophenotyping can be performed on bone marrow biopsy using antibodies to terminal deoxynucleotidyl transferase (TdT), PAX-5 to show B-cell lineage and CD3 to show T-cell lineage (Fig. 19.2B, C, D).
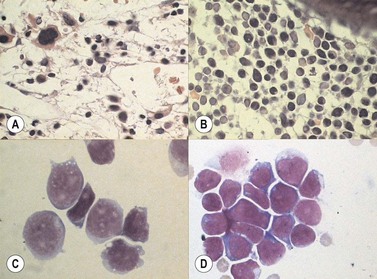
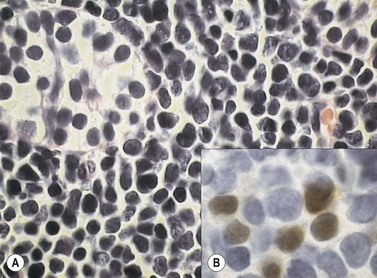
At both presentation or relapse, almost any organ system can be infiltrated by leukemia (Fig. 19.3D). The CNS (Fig. 19.3C) and the testis (Fig. 19.4) are favored sites of relapse.
Immunophenotypic diagnosis
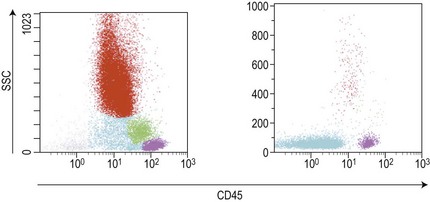
Similar to morphology, the immunophenotypic study of suspected acute leukemia cases is an essential component of diagnosis, allowing further definition of the lineage and maturation stage of blast cells. Immunophenotyping results by flow cytometry can be obtained within a few hours after sampling and are very important in the first stages of therapy initiation. Even if the initial (cytologic) investigations strongly suggest a diagnosis of ALL, a comprehensive immunophenotyping should be carried out, including non-lymphoid lineage markers to exclude undifferentiated acute myeloid leukemia or mixed phenotype acute leukemia (MPAL). Multiparameter flow cytometry on whole BM or PB has now become the standard procedure. A sound strategy should make use of CD45-gating combined with the side scatter (SSC) signal of flow cytometry, which allows to better identify the blast population. In most ALL cases, blasts display low CD45 expression and low SSC characteristics (Fig. 19.5). There is currently no recommended set of immunophenotyping combinations, but a mandatory panel of markers has been proposed by the European Leukemia Net (www.leukemia-net.org). The best orientation combination towards B-lineage, T-lineage or myeloid lineage leukemia relies on the intracytoplasmic detection of relevant antigens. Concomitant labeling, after permeabilization, for cytoplasmic CD79a, CD3 and myeloperoxidase will usually display positivity for only one of these markers, therefore pointing at the relevant lineage. However, in some cases of T-ALL/LBL a weak expression of cytoplasmic CD79a has been reported.10,11 Nuclear labeling for TdT may further differentiate between precursor (positive) and mature (negative) lymphoproliferations (Fig. 19.6). This step should be followed or accompanied by extensive cell surface marker investigation.
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


