Postrenal Failure
- Ureteral obstruction (e.g., calculus, tumor, clot, sloughed papillae, and external compression)
- Bladder outlet obstruction (e.g., prostatic hypertrophy, neurogenic bladder, carcinoma, and urethral stricture)
DIAGNOSIS
Clinical Presentation
History
- Urine history
- Establish urine volume and recent trends.
- Elicit any history of hematuria, proteinuria, dysuria, or pyuria.
- Urgency, frequency, dribbling, and incontinence, especially in elderly men, may direct to prostatic disease.
- Establish urine volume and recent trends.
- Drug history
- Look for nephrotoxins such as NSAIDs, angiotensin-converting enzyme (ACE) inhibitors, angiotensin receptor blockers (ARBs), aminoglycosides, or radiocontrast agents.
- The history should include over-the-counter formulations and herbal remedies or recreational drugs.
- Look for nephrotoxins such as NSAIDs, angiotensin-converting enzyme (ACE) inhibitors, angiotensin receptor blockers (ARBs), aminoglycosides, or radiocontrast agents.
- Volume status
- History of thirst or orthostatic lightheadedness may suggest intravascular depletion.
- Weight gain, ankle swelling, orthopnea, or paroxysmal nocturnal dyspnea may signify fluid retention.
- Look for the possible causes of fluid loss.
- Gastrointestinal: diarrhea, vomiting, and prolonged nasogastric drainage
- Renal: diuretics and osmotic diuresis in hyperglycemia or hypercalcemia
- Dermal: burns and extensive sweating
- Third spacing: acute pancreatitis, ascites, and muscle trauma
- Gastrointestinal: diarrhea, vomiting, and prolonged nasogastric drainage
- History of thirst or orthostatic lightheadedness may suggest intravascular depletion.
- Other potential causes
- Chronic liver disease may cause hepatorenal syndrome.
- Hepatitis C with purpura may suggest cryoglobulinemia.
- Arthralgias, skin rash, and oral ulcers may suggest a connective tissue disorder.
- Sinusitis, cough, and hemoptysis may alert the physician to the possibility of granulomatosis with polyangiitis (formerly Wegener granulomatosis) or Goodpasture syndrome.
- A history of recent sore throat or significant skin infection may suggest acute poststreptococcal glomerulonephritis.
- Low back pain and anemia may suggest multiple myeloma.
- Myalgias, dark-colored urine, and the appropriate clinical scenario (crush injury, exercise, immobilization) may suggest rhabdomyolysis.
- Chronic liver disease may cause hepatorenal syndrome.
Physical Examination
- Orthostatic vital signs, mucous membrane and skin turgor, and examination of jugular veins can assess the patient’s fluid balance. Looking for sacral edema in a supinepositioned patient is also important.
- The presence of an S3, pulmonary crackles, and pitting edema suggests volume overload.
- The presence of an abdominal bruit suggests renovascular disease.
- Pelvic examination in females and rectal examination in both females and males may detect a cause of postrenal obstruction.
- The kidney can be palpable in cases of hydronephrosis or polycystic kidney disease.
Diagnostic Testing
Laboratories
- Routine urinalysis and microscopic analysis of urine should always be done and are often helpful in determining the cause of AKI. Usually, the urine is bland in prerenal and uncomplicated postrenal AKI, while an abnormal urinalysis and active sediment suggest an intrinsic renal cause.
- Prerenal AKI: usually bland, with occasional hyaline casts
- ATN: muddy brown granular casts, epithelial cells, and epithelial cell casts
- Glomerulonephritis: dysmorphic red blood cells (RBCs) and RBC casts
- Acute interstitial nephritis: eosinophils, other white blood cells (WBCs), and WBC casts
- Prerenal AKI: usually bland, with occasional hyaline casts
- In prerenal states, tubular function is intact, and the kidney avidly retains sodium, usually resulting in low urine sodium and a fractional excretion of sodium (FENa) of <1%. FENa is particularly helpful in oliguric AKI (Table 24-2).
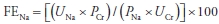
where U = urine, P = plasma, Na = sodium, and Cr = creatinine.
- Because loop diuretics force natriuresis, calculation of FENa is misleading in patients who are taking these agents. The fractional excretion of urea (FEurea) of <30% to 35% is suggestive of prerenal azotemia (Table 24-2).
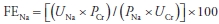
where Uurea = urine urea, P = plasma, BUN = blood urea nitrogen (mg/dL), and Cr = creatinine.
TABLE 24-2 Laboratory Tests in the Differentiation of Oliguric Prerenal Azotemia from Oliguric Intrinsic Acute Tubular Necrosis

FENa, fractional excretion of sodium; Plasma BUN/Cr, plasma blood urea nitrogen to creatinine ratio; UNa, urine sodium concentration; Uosm, urine osmolality; U/PCr, urine to plasma creatinine ratio.
Imaging
- Ultrasonography exhibits high sensitivity (90% to 98%) but a lower specificity (65% to 84%) for the detection of urinary tract obstruction.5 It can also measure the echotexture (increased echogenicity suggests more chronic damage) and kidney size (a marked difference may suggest renovascular disease).
- Compared with renal ultrasonography, noncontrast CT is superior in the evaluation of ureteral obstruction since it can define the level of obstruction and demarcate retroperitoneal fibrosis or a retroperitoneal mass.
Diagnostic Procedures
Renal biopsy is reserved for patients in whom the cause of intrinsic AKI is unclear or in cases of unexplained proteinuria or hematuria where immunosuppressive therapy is being considered.
TREATMENT
Nondialytic Therapy
- Volume expansion: Prompt and effective restoration of effective circulating volume is the key in prerenal azotemia due to true hypovolemia.
- Avoidance of nephrotoxins: Contrast media should be avoided when possible. ACE inhibitors, ARBs, diuretics, and NSAIDs should be held when there is a sudden decline in renal function.
- Electrolyte management: Hyperkalemia is a potentially lethal complication of AKI. Hyperphosphatemia and hypocalcemia are also common in AKI.
- Rule out pseudohyperkalemia by repeating a whole-blood potassium. Consider drawing the sample without the use of a tourniquet or fist clenching.
- If the patient has thrombocytosis or marked leukocytosis, the sample may be drawn in a heparinized tube.
- Obtain a stat ECG and arterial blood gases (ABG) if acidosis is a concern.
- Review the patient’s medication list and stop all exogenous K+ and potentially offending drugs.
- Acute treatment is necessary for severe hyperkalemia.
- Calcium gluconate 10%, 10 mL IV over 2 to 3 minutes, decreases cardiac membrane excitability but does not alter the potassium concentration. The effect occurs in minutes but lasts only 30 to 60 minutes. It can be repeated after 5 to 10 minutes if the ECG does not change. Use with extreme caution in patients receiving digoxin.
- Insulin, 10 units of regular insulin IV, causes an intracellular shift of K+ in 10 to 30 minutes. The effect lasts for several hours. Glucose, 50 g IV (2 ampules D50), should be administered concurrently to prevent hypoglycemia.
- β2-Adrenergic agonists can be used to cause an intracellular shift of K+.
- Diuretics (e.g., furosemide 40 to 120 mg IV) enhance K+ excretion provided renal function is adequate.
- Cation exchange resins (sodium polystyrene sulfonate, Kayexalate) enhance K+ excretion from the gastrointestinal tract. Kayexalate may be given PO (15 to 30 g) or as a retention enema (30 to 50 g). The effect may not be evident for several hours and lasts 4 to 6 hours. Doses may be repeated every 4 to 6 hours as needed. The oral preparation is preferred given reports of intestinal necrosis in select patients after rectal administration.
- NaHCO3, 1 ampule (50 mEq) IV, can also be used to cause an intracellular shift of K+, and the effect can last several hours. Its effect is minimal in organic acidoses. Patients with end-stage renal disease (ESRD) seldom respond and may not tolerate the Na+ load.
- Dialysis may be necessary for severe hyperkalemia when other measures are ineffective and for patients with renal failure.
- Rule out pseudohyperkalemia by repeating a whole-blood potassium. Consider drawing the sample without the use of a tourniquet or fist clenching.
- Acid-base disorders: Metabolic acidosis is a common complication of AKI. If severe, alkali therapy may be required.
- Severe acidosis (pH < 7.20) may require treatment with parenteral NaHCO3. Rapid infusion should be considered only for very severe acidosis.
- The bicarbonate deficit may be estimated as follows:
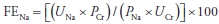
- Overaggressive correction should be avoided to prevent overshoot alkalosis.
- Hypernatremia and fluid overload can occur with NaHCO3 administration.
- Serum electrolytes, including calcium, should be followed closely.
- Severe acidosis (pH < 7.20) may require treatment with parenteral NaHCO3. Rapid infusion should be considered only for very severe acidosis.
- Nutrition support is an important facet of conservative care.
- Treatment for specific causes of AKI
- Immunosuppressive agents for glomerulonephritis or vasculitis
- Systemic anticoagulation for renal artery or vein thrombosis
- Plasmapheresis for hemolytic-uremic syndrome (HUS)/thrombotic thrombocytopenic purpura
- Immunosuppressive agents for glomerulonephritis or vasculitis
Dialytic Therapy for Acute Renal Failure
Indications for initiation of dialytic support in acute renal failure include the following:
- Severe hyperkalemia, metabolic acidosis, or volume overload refractory to medical therapy
- Uremic syndrome with uremic pericarditis, encephalopathy, or seizures
- Need to start total parental nutrition (volume/solute issues)
- Overdose/intoxications
- Refractory hypercalcemia
- Refractory hyperuricemia
Glomerulopathy
GENERAL PRINCIPLES
- Glomerular diseases are manifestations of primary kidney pathology or representations of kidney involvement of a multisystem disease.
- The etiology of many glomerular diseases remains unknown.
- Glomerular disease may be asymptomatic and found on routine patient evaluation for systemic diseases or in patients noted to have hypertension (HTN), edema, proteinuria, and/or hematuria (Fig. 24-1).1
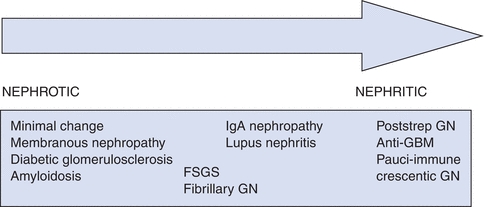
Figure 24-1 Spectrum of glomerular diseases with nephrotic and nephritic features. FSGS, focal segmental glomerulosclerosis; GBM, glomerular basement membrane; GN, glomerulonephritis; strep, streptococcal. (From Khalid SA. Overview and approach to the patient with glomerular disease. In: Cheng S, Vijayan A, eds. The Washington Manual Nephrology Subspecialty Consult, 3rd ed. Philadelphia, PA: Lippincott Williams & Wilkins; 2012:192.)
DIAGNOSIS
Clinical Presentation
- The presentation of some glomerulopathies can be asymptomatic.
- Isolated proteinuria: A daily protein excretion of >150 mg is abnormal. When proteinuria exceeds 3.5 g/day, it is termed nephrotic range and is highly likely to be caused by a glomerular lesion. Microalbuminuria is excretion of 30 to 300 mg albumin per day and is not typically detected on routine urine dipstick. This test is commonly used in diabetic subjects to help identify patients at risk of developing nephropathy.
- Isolated hematuria is classified as either microscopic, referring to more than two RBCs per high power field (HPF) in spun urine, or macroscopic, referring to visible tea-colored (brown/red) urine. In patients with glomerular disease, macroscopic hematuria is usually not associated with pain. The presence of dysmorphic RBCs and/or RBC casts in the urine sediment would be highly suggestive of a glomerular source of hematuria.
- The presence of proteinuria with hematuria is a stronger indicator of an underlying glomerular disease process.
- Isolated proteinuria: A daily protein excretion of >150 mg is abnormal. When proteinuria exceeds 3.5 g/day, it is termed nephrotic range and is highly likely to be caused by a glomerular lesion. Microalbuminuria is excretion of 30 to 300 mg albumin per day and is not typically detected on routine urine dipstick. This test is commonly used in diabetic subjects to help identify patients at risk of developing nephropathy.
- The nephritic syndrome presents with proteinuria <3.5 g/day, edema, hematuria with dysmorphic RBC and/or casts, renal failure with or without oliguria, and HTN.
- The nephrotic syndrome is characterized by presence of proteinuria >3.5 g/day, hypoalbuminemia <3.5 g/dL, hyperlipidemia, lipiduria, and edema. It is associated with an increased risk of:
- Atherosclerosis and hyperlipidemia.
- Thromboembolic events due to underlying hypercoagulability secondary to urinary loss of antithrombotic proteins, including proteins C and S, and antithrombin III. Venous thromboembolic disease is more common than arterial thrombosis.
- Infection due to urinary loss of immunoglobulins.
- Atherosclerosis and hyperlipidemia.
- Rapidly progressive glomerulonephritis (RPGN) is an acute presentation of glomerular injury leading to renal failure, which develops in days to weeks. This is a severe manifestation of the nephritic syndrome. The key pathologic finding in RPGN is formation of cellular crescents within the Bowman space. When this involves more than 50% of the glomeruli, it is commonly referred to as crescentic GN.
- Chronic glomerulonephritis is a slowly progressive glomerular disease that leads to renal failure over a period of months to years. It is suggested by HTN, proteinuria >3 g/day, chronic renal insufficiency, and small atrophic kidneys.
History
- Ask about symptoms like joint pain, rash, peripheral and/or periorbital edema, foamy urine, and hematuria. Patients with RPGN may have in addition symptoms of fever, nose bleeds, hemoptysis or vague symptoms of hair loss, fatigue, and weight loss.
- Past history focuses on history of malignancies and infections. Timing of infection can be very helpful. IgA nephropathy will have hematuria within 1 to 3 days after onset of respiratory symptoms as opposed to postinfectious GN, which develops 1 to 3 weeks after a streptococcal upper respiratory tract infection and 5 to 6 weeks after a skin infection.
- Some GN have a familial nature like Alport syndrome (renal failure associated with hearing loss), thin basement membrane disease, IgA nephropathy, focal segmental glomerulosclerosis (FSGS), and atypical HUS.
- Medication use including prescription, over-the-counter medications, and herbal supplements should be evaluated. Many drugs and toxins may be associated with glomerular disease.
- NSAIDs are associated with minimal change disease.
- Penicillamine, gold, NSAIDs, and mercury are associated with membranous nephropathy.
- Heroin has previously been associated with FSGS.
- Cyclosporine, tacrolimus, and mitomycin C are associated with thrombotic microangiopathy.
- Various malignancies are associated with glomerular disease.
- NSAIDs are associated with minimal change disease.
- Review of systems should cover multisystem diseases associated with glomerular disease such as HTN, diabetes mellitus (DM), systemic lupus erythematosus (SLE), amyloid, and vasculitis.
Physical Examination
- Periorbital edema in the mornings is a highly suggestive feature of the nephrotic syndrome. Facial edema is most often absent in heart failure or in patients with liver cirrhosis due to the inability of these patients to lie flat.
- Xanthelasmas may be present due to hyperlipidemia associated with nephrotic syndrome.
- Muehrcke bands (white bands in fingernails parallel to the lunula) may also be present from hypoalbuminemia in patients with nephrotic syndrome.8
- Palpable purpura may be seen in cryoglobulinemia, vasculitis, or SLE.
- Malar rash can be seen in patients with SLE.
- Evaluation for piercing and skin tattoos may give a clue to underlying hepatitis or HIV, both of which can be associated with glomerular disease.
Differential Diagnosis
Minimal Change Disease
- Minimal change disease (MCD) is defined by the presence of nephrotic syndrome, the absence of histologic glomerular abnormality by light microscopy, and evidence of podocyte foot process effacement by electron microscopy.
- Most commonly presents in children between ages of 2 and 7 years, but the disease may occur at any age.
- Most cases are idiopathic. However, in a number of patients, the onset is preceded by allergic reaction, vaccination, or viral infection. Around 20% of the patients have a history of atopy.9 Secondary causes include the following:
- Drugs: NSAIDs, lithium, interferon-α
- Infections: HIV, syphilis
- Malignancies: Hodgkin lymphoma, rarely non-Hodgkin lymphoma, and solid tumors
- Drugs: NSAIDs, lithium, interferon-α
- Onset of nephrotic syndrome is typically abrupt. Creatinine is generally normal at presentation but can be elevated in some adults.
- Urine sediment is usually bland. Complement levels are normal. Renal biopsy is required for diagnosis. Glomeruli appear normal on light microscopy, with no immunoglobulin or complement deposition on immunofluorescence. Presence of foot process effacement is seen on electron microscopy.
- Complications of MCD include infection, peritonitis, thromboembolism, and acute renal failure, especially in the setting of hypovolemia.
Focal and Segmental Glomerulosclerosis
- FSGS is defined by glomerular lesions characterized by segments of sclerosis in only a portion (segmental) of some glomeruli (focal). FSGS has become an important form of glomerular disease in the US due to its increasing cause of ESRD.10 It can be classified as primary or secondary FSGS.
- Primary idiopathic FSGS
- The most common cause of idiopathic nephrotic syndrome in adults. The disease is markedly more common in African Americans.
- Pathogenesis remains unclear, but the presence of a circulating permeability factor called soluble urokinase plasminogen activator receptor (suPAR) has been seen in nearly 60% to 80% of adult patients with primary FSGS.11 Circulating suPAR affects podocyte maturation and function thereby leading to damage at the glomerular filtration barrier.
- FSGS presents most often as nephrotic syndrome but may also present in 20% to 30% as persistent nonnephrotic range proteinuria, HTN, microscopic hematuria, and renal insufficiency.
- There is frequent progression to ESRD, up to 50% at 10 years.
- Diagnosis requires renal biopsy. Multiple histologic variants of FSGS have been described. The so-called tip lesion variant has a better prognosis, while the collapsing variant is associated with worse outcomes.
- The most common cause of idiopathic nephrotic syndrome in adults. The disease is markedly more common in African Americans.
- Secondary FSGS occurs as an adaptive response to glomerular hyperfiltration or podocyte injury. Podocyte injury can be caused by many factors like viral infection, drugs, or genetic mutation affecting the structural integrity of the podocytes.
- HIV-associated nephropathy: Up to 95% of cases occur in African Americans. Renal ultrasound shows enlarged kidneys with increased echogenicity. The collapsing form of FSGS is most notably associated with HIV.
- Reduced renal mass (unilateral renal agenesis, renal ablation, and renal allograft): This initially leads to an adaptive hyperfiltration, which over time causes glomerular HTN and FSGS.
- Hypoxemia in the setting of sickle cell anemia, congenital pulmonary disease, or cyanotic congenital heart disease leads to glomerular enlargement and subsequently causes FSGS.
- Toxins from impurities in heroin have been known to cause podocyte damage and an FSGS pattern.
- Chronic vesicoureteral reflux.
- Morbid obesity.
- Genetic forms of FSGS occur due to mutations in genes affecting podocyte and slit diaphragm structures. Variation in the APOL1 gene, (codes for apolipoprotein L-1), a protective mechanism against Trypanosoma brucei brucei, has been associated with FSGS in the presence of HTN among African Americans.12
- HIV-associated nephropathy: Up to 95% of cases occur in African Americans. Renal ultrasound shows enlarged kidneys with increased echogenicity. The collapsing form of FSGS is most notably associated with HIV.
Membranous Nephropathy
- Membranous nephropathy (MN) is a glomerular disease characterized by subepithelial immune deposits of IgG and complement along the glomerular basement membrane with characteristic spike pattern seen on methenamine silver staining.
- MN represents the most common cause of idiopathic nephrotic syndrome in adults >60 years of age and the second most common cause (after FSGS) of the nephrotic syndrome in all adults.
- The pathogenesis of MN remains unclear, but recent experiments have identified circulating antibodies to the M-type phospholipase A2 receptor (anti-PLA2R antibody) in 70% of patients with idiopathic MN.13 Another antibody targeting neutral endopeptidase (NEP) has been attributed to the development of congenital MN. Secondary causes of MN include the following:
- Malignancies (colon, kidney, lung) account for the majority of secondary MN. Nephrotic syndrome may precede clinical manifestations of malignancy by up to 2 years.1
- Autoimmune diseases including SLE, type 1 DM, rheumatoid arthritis, and myasthenia gravis.
- Infections such as hepatitis B, hepatitis C, and syphilis.
- Drugs including NSAIDs, gold, and penicillamine.
- Malignancies (colon, kidney, lung) account for the majority of secondary MN. Nephrotic syndrome may precede clinical manifestations of malignancy by up to 2 years.1
- About 80% of patients with MN present with overt nephrotic syndrome. Microscopic hematuria may be seen in up to 50% of adults. Serum complement levels are normal in idiopathic MN.
- Deep vein thrombosis, especially renal vein thrombosis, is more common in MN than in other forms of nephrotic syndrome.
Membranoproliferative Glomerulonephritis
- Histopathologically, membranoproliferative glomerulonephritis (MPGN) is characterized by diffuse mesangial proliferation, thickening of the capillary loops due to reduplication of glomerular capillary basement membrane (double-contour or tram track), subendothelial immune deposits, and mesangial hypercellularity. Most cases are associated with circulating immune complexes and low serum complements.9
- Based on the histomorphologic pattern, there are three types of MPGN.14
- Type 1: discrete immune deposits in the mesangium and subendothelial space. This is frequently associated with hepatitis C infection, cryoglobulinemia, or endocarditis.
- Type 2: continuous, intramembranous, ribbon-like deposits. It is often called dense-deposit disease. This can be associated with partial lipodystrophy, C3 nephritic factor, and factor H or I deficiency with complement consumption.9
- Type 3: diffuse subepithelial and subendothelial deposits within the glomerular basement membrane.
- Type 1: discrete immune deposits in the mesangium and subendothelial space. This is frequently associated with hepatitis C infection, cryoglobulinemia, or endocarditis.
- MPGN can manifest as microscopic hematuria and proteinuria, nephritic syndrome, nephrotic syndrome, or chronic progressive GN. The majority of patients have HTN.
- MPGN almost always is associated with low serum C3 and/or C4 levels.
- Primary MPGN is a diagnosis of exclusion after potential secondary causes like hepatitis infection, HIV/AIDS, infective endocarditis, collagen vascular disease, malignancy, or chronic liver disease have been excluded.
IgA Nephropathy and Henoch-Schönlein Purpura
- IgA nephropathy (also known as Berger disease) is a mesangial proliferative GN characterized by diffuse mesangial deposition of IgA.
- IgA nephropathy is the most common primary GN globally.
- There is a higher incidence in Asia and Europe, which may be due to difference in biopsy practices for isolated microscopic hematuria.
- Pathogenesis of IgA nephropathy remains unclear. Half of the patients have a preceding infection. There is reduced galactosylation of the O-linked hinge region sugar of circulating IgA1. This leads to production of IgG antibodies against the circulating abnormal IgA predisposing to formation of immune complexes and mesangial deposition.15
- IgA nephropathy can present as episodic hematuria, asymptomatic microscopic hematuria with variable degrees of proteinuria, or uncommonly as a rapidly progressive crescentic disease. Hematuria is seen within 1 to 3 days following upper respiratory infection. This distinguishes IgA nephropathy from poststreptococcal GN, in which the hematuria is delayed by 1 to 3 weeks.
- ESRD develops in 25% to 30% of patients within 20 years of diagnosis. Presence of uncontrolled HTN, persistent proteinuria >1 g/day, impaired renal function, and older age at diagnosis are risk factors for progression.16
- Approximately 30% to 50% of patients have increased serum IgA levels, but levels do not correlate with disease activity. Complement levels are usually normal.
- Henoch-Schönlein purpura is a syndrome with IgA nephropathy and systemic small vessel vasculitis caused by IgA deposits. It presents with arthralgia, purpuric skin rash, abdominal pain, or gastrointestinal bleeding. It predominantly affects children.
Glomerulonephritis in Multisystem Disorders
- Systemic lupus erythematosus
- Lupus nephritis is an immune complex–mediated complication of SLE that presents with various histologic patterns described by the 2003 International Society of Nephrology and Renal Pathology Society, based on the degree of glomerular involvement (Table 24-3).
- Renal biopsy plays a critical role in diagnosis and management of patients with lupus nephritis but remains a topic of controversy in predicting outcomes and prognosis.17 Classically, the proliferative forms of SLE (Classes III and IV) are associated with an increased risk of renal failure.
- About 50% to 60% of patients with lupus have renal involvement during their disease course. Lupus nephritis may manifest as benign asymptomatic hematuria, proteinuria, or even fulminant renal failure.1
- Serum complement levels are usually low in the majority of patients because of classic complement pathway activation.18
- Lupus nephritis is an immune complex–mediated complication of SLE that presents with various histologic patterns described by the 2003 International Society of Nephrology and Renal Pathology Society, based on the degree of glomerular involvement (Table 24-3).
- Pauci-immune glomerulonephritis is a group of small vessel vasculitides characterized by the presence of necrotizing crescents with minimal or no immune complex deposits.
- The majority (80%) of the patients have positive circulating antineutrophil cytoplasmic antibodies (ANCA) and hence are also called ANCA-associated vasculitis.
- Pauci-immune GN includes granulomatosis with polyangiitis (GPA, formerly called as Wegener granulomatosis), microscopic polyangiitis (MPA), eosinophilic granulomatosis with polyangiitis (EGPA, formerly called Churg-Strauss syndrome), and renal limited vasculitis.19
- Patients can present with nonspecific symptoms of malaise, weight loss, fever, and arthralgia.
- Renal manifestations include hematuria, varying degrees of proteinuria, and dysmorphic RBCs with RBC casts.
- Disease-specific manifestations:
- GPA: sinusitis, nasopharyngeal mucosal ulceration, hemoptysis, purpura, and renal involvement. C-ANCA and proteinase 3 (PR3) are positive in 65% to 90% of patients with active disease. Serum complement levels are normal.
- EGPA: asthma, peripheral eosinophilia, and renal involvement. P-ANCA and myeloperoxidase (MPO) are associated with EGPA.
- MPA: generally associated with renal involvement and absence of granulomas.
- GPA: sinusitis, nasopharyngeal mucosal ulceration, hemoptysis, purpura, and renal involvement. C-ANCA and proteinase 3 (PR3) are positive in 65% to 90% of patients with active disease. Serum complement levels are normal.
- The majority (80%) of the patients have positive circulating antineutrophil cytoplasmic antibodies (ANCA) and hence are also called ANCA-associated vasculitis.
- Goodpasture disease is suggested by the presence of antiglomerular basement membrane (anti-GBM) antibody along with a clinical presentation of pulmonary-renal syndrome consisting of hemoptysis, pulmonary infiltrates, and/or RPGN.
- Anti-GBM antibodies are directed against the noncollagenous-1 domain of the α-3 chain of type IV collagen.
- This disease is characterized by focal necrotizing crescentic GN in association with circulating anti-GBM antibodies in the blood and linear staining of IgG along the glomerular basement membrane.
- Approximately 30% of patients with Goodpasture disease are P-ANCA positive.
- Anti-GBM antibodies are directed against the noncollagenous-1 domain of the α-3 chain of type IV collagen.
- Poststreptococcal glomerulonephritis (PSGN) is an immune complex–mediated GN characterized by renal deposition of complement C3 and IgG as subepithelial humps on electron microscopy. It is associated with low serum C3 and CH50 and occurs following an infection with nephritogenic strains of group A or, sometimes, group C streptococci.
- PSGN is principally a disease of children, but has been increasingly reported in older patients occurring 1 to 3 weeks after pharyngitis or 5 to 6 weeks after impetigo. Incidence of PSGN has progressively declined in the industrialized countries.
- Clinical presentation consists of hematuria manifesting as tea-colored urine, edema, HTN, and oliguria. Prognosis is excellent in children and less favorable in adults.
- Laboratory tests include antibodies to streptococcal antigens (antistreptolysin-O and anti-DNase B) and hypocomplementemia.
- Atypical postinfectious GN is increasingly reported following staphylococcal and gram-negative infections. It is more commonly seen in patients with underlying diabetes. Clinical presentation is more severe than PSGN with a greater number of patients progressing to ESRD. Predominance of IgA deposition and C3 can be seen on renal biopsy with no subepithelial humps.9
- PSGN is principally a disease of children, but has been increasingly reported in older patients occurring 1 to 3 weeks after pharyngitis or 5 to 6 weeks after impetigo. Incidence of PSGN has progressively declined in the industrialized countries.
- Thrombotic microangiopathies (TMA) is a syndrome characterized by microangiopathic hemolytic anemia (MAHA), thrombocytopenia, and varying degrees of organ dysfunction.
- TMA can be seen in a variety of clinical conditions including thrombotic thrombocytopenic purpura (TTP), HUS, malignant HTN, scleroderma, antiphospholipid syndrome (APLA), HIV infection, and secondary to medications like calcineurin inhibitors and clopidogrel.
- Diagnostic criteria for TTP include MAHA, thrombocytopenia, fever, neurologic signs, and renal failure. Low circulating levels or antibodies against ADAMTS13 lead to increased circulating ultralarge vWF (ULvWF) consequently causing platelet activation and aggregation.
- HUS is classically associated with diarrheal infection caused by exotoxins produced by Escherichia coli (O157:H7 serotype) or Shigella dysenteriae type 1.
- Atypical HUS (aHUS) is caused by dysregulation of the complement system.20 Absence of diarrhea and leukocytosis with clinical features of HUS and hypocomplementemia should raise the suspicion of aHUS.9
- TMA can be seen in a variety of clinical conditions including thrombotic thrombocytopenic purpura (TTP), HUS, malignant HTN, scleroderma, antiphospholipid syndrome (APLA), HIV infection, and secondary to medications like calcineurin inhibitors and clopidogrel.
- Amyloidosis is characterized by the deposition of extracellular, insoluble, polymeric protein fibrils.
- Classification is based on the type of protein precursor that forms the amyloid fibril. Renal involvement is most often seen in primary amyloidosis (AL) and systemic secondary (AA) amyloidosis.
- Protein precursor for AL amyloid is light chain and hence can be seen in patients with multiple myeloma.
- Clinical features include proteinuria with or without microscopic hematuria and renal insufficiency. Monoclonal light chains can be detected by measuring serum free light chains (SFLC) and serum or urine protein electrophoresis with immunofixation.21
- Congo red stain will show an apple-green birefringence if AL amyloid is present on biopsy specimen.
- Prognosis of AL amyloidosis is poor with a median survival of <2 years.
- Classification is based on the type of protein precursor that forms the amyloid fibril. Renal involvement is most often seen in primary amyloidosis (AL) and systemic secondary (AA) amyloidosis.
TABLE 24-3 Abbreviated (ISN/RPS) Classification of Lupus Nephritis (2003)

Indicate and grade (mild, moderate, severe) tubular atrophy, interstitial inflammation and fibrosis, severity of arteriosclerosis, or other vascular lesions.
a Indicate the proportion of glomeruli with active and with sclerotic lesions.
b Indicate the proportion of glomeruli with fibrinoid necrosis and cellular crescents.
c Class V may occur in combination with class III or IV in which case both will be diagnosed.
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


