17 Acute kidney injury
Definition and incidence
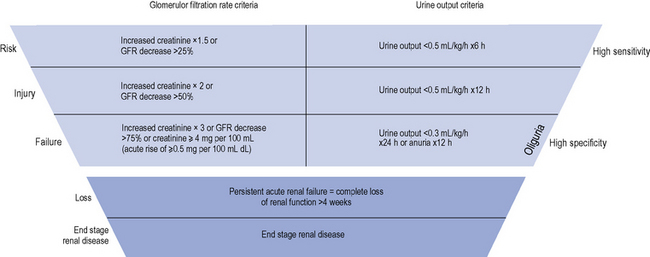
The diagnostic criteria for AKI is based on an increase in serum creatinine or the presence of oliguria (see Table 17.1). Criteria have recently been introduced for the definition and staging of the condition; the acronym RIFLE is used (Risk, Injury, Failure, Loss and End-stage renal disease (ESRD)), which is now becoming established in clinical practice (see Fig. 17.1).
Table 17.1 Classification of acute kidney injury
| Acute kidney injury type | Typical % cases | Common aetiology |
|---|---|---|
| Pre-renal | 40–80 | Reversible ↓ renal perfusion through hypoperfusion |
| Intra-renal (including ATN) | 10–50 | Renal parenchymal injury |
| Post-renal | <10 | Urinary tract obstruction |
ATN, acute tubular necrosis.
Classification and causes
AKI is not a single disease state with a uniform aetiology, but a consequence of a range of different diseases and conditions. The most useful practical classification comprises three main groupings: (i) pre-renal, (ii) renal, or (iii) post-renal. More than one category may be present in an individual patient. Common causes of each type of AKI are outlined in Table 17.1.
Pre-renal acute kidney injury
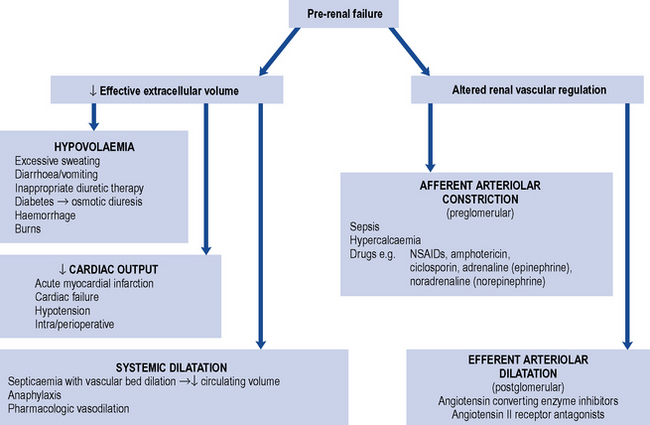
This is caused by impaired perfusion of the kidneys with blood, and is usually a consequence of decreased intravascular volumes (hypovolaemia) and/or decreased intravascular pressures. Some of the commonest causes of pre-renal AKI are summarised in Fig. 17.2. Perfusion of the kidneys at the level of the microvascular beds (glomerular and tubulo-interstitial) is usually maintained through wide variations in pressure and flow through highly efficient auto-regulatory pathways, such as the renin–angiotensin–aldosterone system (RAAS) and regulated prostaglandin synthesis. However, when the systolic blood pressure (BP) drops below 80 mmHg, AKI may develop. In individuals with chronic kidney disease (CKD) or in the elderly, this may occur at higher levels of systolic BP. Drugs that inhibit the RAAS, such as angiotension converting enzyme inhibitors (ACE inhibitors) and angiotensin receptor blockers (ARBs), or block the production of prostaglandins, such as non-steroidal anti-inflammatory drugs (NSAIDs), can pre-dispose to the development of pre-renal AKI. These are discussed in more detail below.
Intra-renal acute kidney injury
This is caused by a variety of causes (see Tables 17.1 and 17.2), most commonly (in >80% of cases) acute tubular necrosis (ATN). ATN occurs usually as a consequence of a combination of factors, including hypotension, often in the setting of sepsis and nephrotoxic agents including drugs or chemical poisons, or endogenous sources such as myoglobin or haemoglobin.
Table 17.2 Common clinical factors known to cause acute tubular necrosis
| Clinical factor | Mechanism |
|---|---|
| Hypoperfusion | Reduced oxygen/nutrient supply |
| Radiocontrast media | Medullary ischaemia may result from contrast media induced renal vasoconstriction. The high ionic load of contrast media may produce ischaemia particularly in diabetics and those with myeloma (who produce large quantities of light chain immunoglogulins) |
| Sepsis | Infection produces endotoxaemia and systemic inflammation in combination with a pre-renal state and nephrotoxins. The immunological response to sepsis involves release of vasoconstrictors and vasodilators (e.g. eicosanoids, nitric oxide) and damage to vascular endothelium with resultant thrombosis |
| Rhabdomyolysis | Damaged muscles release myoglobin, which can cause ATN through direct nephrotoxicity and by a reduction in blood flow in the outer medulla |
| Renal transplantation | The procedures and conditions encountered during renal transplantation can induce ischaemic ATN which can be difficult to distinguish from the nephrotoxic effects of immunosuppressive drug therapy used in these circumstances and rejection |
| Hepatorenal syndrome | Renal vasoconstriction is frequently seen in patients with end-stage liver disease. Progression to ATN is common |
| Nephrotoxins | |
| Aminoglycosides | Aminoglycosides are transported into tubular cells where they exert a direct nephrotoxic effect. Current dosage regimens recommend once daily doses, with frequent monitoring of drug levels, to minimise total uptake of aminoglycoside |
| Amphotericin | Amphotericin appears to cause direct nephrotoxicity by disturbing the permeability of tubular cells. The nephrotoxic effect is dose dependent and minimised by limiting total dose used, rate of infusion and by volume loading. These precautions also apply to newer liposomal formulations |
| Immunosuppressants | Ciclosporin and tacrolimus cause intra-renal vasoconstriction that may result in ischaemic ATN. The mechanism is unclear but enhanced by hypovolaemia and other nephrotoxic drugs |
| NSAIDs | Vasodilator prostaglandins, mainly E2, D2 and I2 (prostacyclin), produce an increase in blood flow to the glomerulus and medulla. In normal circumstances, they play no part in the maintenance of the renal circulation. However, increased amounts of vasoconstrictor substances arise in a variety of clinical conditions such as volume depletion, congestive cardiac failure or hepatic cirrhosis associated with ascites. Maintenance of renal blood flow then becomes more reliant on the release of vasodilatory prostaglandins. Inhibition of prostaglandin synthesis by NSAIDs may cause unopposed arteriolar vasoconstriction, leading to renal hypoperfusion |
| Cytotoxic chemotherapy | For example, cisplatin |
| Anaesthetic agents | Methoxyflurane, enflurane |
| Chemical poisons/naturally occurring poisons | Insecticides, herbicides, alkaloids from plants and fungi, reptile venoms |
Acute tubular necrosis
ATN is a diagnosis made by renal biopsy; the findings can include damage to the proximal tubule and the ascending limb of the loop of Henle, interstitial oedema and sparse infiltrating inflammatory cells. Whilst severe and sustained hypoperfusion can lead to ATN, it usually develops when there is a combination of factors including the presence of one or more of a range of nephrotoxins. These may arise exogenously from drugs or chemical poisons, or from endogenous sources such as haemoglobin, myoglobin, crystals (uric acid, phosphate) and toxic products from sepsis or tumours (see Table 17.2). Some endogenous toxins may be released as a direct consequence of drug exposure. For example, myoglobin may be released (rhabdomyolosis) following muscle injury or necrosis, hypoxia, infection or following drug treatment, for example, with fibrates and statins, particularly when both are used in combination. The mechanism of the subsequent damage to renal tissue is not understood fully but probably results from a combination of factors including hypoperfusion, haem-catalysed free radical tubular cytotoxicity and haem cast formation and precipitation leading to tubular injury.
Differentiating pre-renal from renal acute kidney injury
It is sometimes possible to distinguish between cases of pre-renal and renal AKI through examination of biochemical markers (see Table 17.3). In renal AKI, the kidneys are generally unable to retain Na+ owing to tubular damage. This can be demonstrated by calculating the fractional excretion of sodium (FENa); in practice this is not often done because it lacks sensitivity and specificity and may be difficult to interpret in the elderly who may have pre-existing concentrating defects.
Table 17.3 Differentiating pre-renal from renal acute kidney injury
| Laboratory test | Pre-renal | Renal |
|---|---|---|
| Urine osmolality (mOsm/kg) | >500 | <400 |
| Urine sodium (mEq/L) | <20 | >40 |
| Urine/serum creatinine (μmol/L) | >40 | <20 |
| Urine/serum urea (μmol/L) | >8 | <3 |
| Fractional excretion of sodium (%) | <1 | >2 |
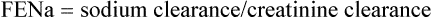
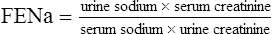
Clinical manifestations
Acute kidney injury with volume depletion
In those patients with volume depletion, a classic pathophysiological picture is likely to be present, with tachycardia, postural hypotension, reduced skin turgor and cold extremities (see Table 17.4). The most common sign in AKI is oliguria, where urine production falls to less than 0.5 mL/kg/h for several hours. This is below the volume of urine required to effectively excrete products of metabolism to maintain a physiological steady state. Therefore, the serum concentration of those substances normally excreted by the kidney will rise and differentially applies to all molecules up to a molecular weight of around 50 kDa. This includes serum creatinine, which at a molecular weight of 113 Da is normally freely filtered by the kidneys but with loss of kidney function the serum level climbs. Whilst the term uraemia is still in widespread use, it merely describes a surrogate for the overall metabolic disturbances that accompany AKI; these include excess potassium, hydrogen ions (acidosis) and phosphate in blood. Most cases of AKI are first identified by an abnormal blood test, though some patients may have symptoms that are specifically attributable to AKI; these include nausea, vomiting, diarrhoea, gastro-intestinal haemorrhage, muscle cramps and a declining level of consciousness.
Table 17.4 Factors associated with acute kidney injury
| Volume depletion | Volume overload | |
|---|---|---|
| History | Thirst | Weight increase |
| Excessive fluid loss (vomiting or diarrhoea) | Orthopnoea/nocturnal dyspnoea | |
| Oliguria | ||
| Physical examination | Dry mucosae ↓ Skin elasticity | Ankle swelling Oedema |
| Tachycardia | Jugular venous distension | |
| ↓ Blood pressure | Pulmonary crackles | |
| ↓ Jugular venous pressure | Pleural effusion |
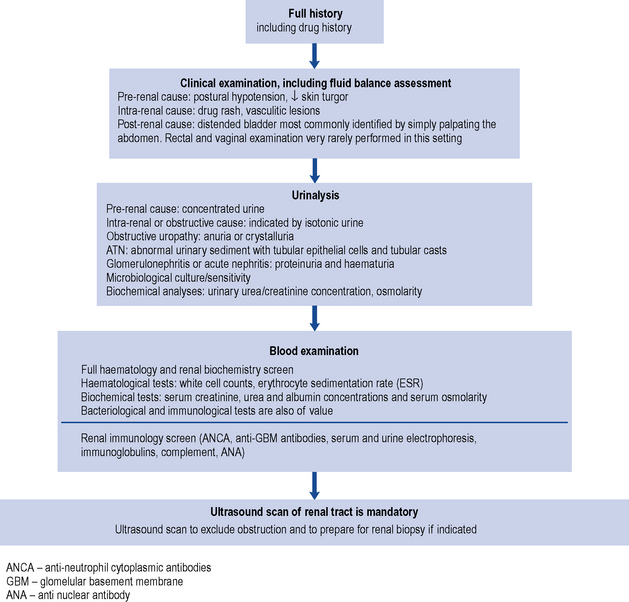
Diagnosis and clinical evaluation
The assessment of renal function is described in detail in Chapter 18. However, unless a patient is at steady state, measurement of serum creatinine does not provide a reliable guide to renal function. For example, serum creatinine levels will usually rise by only 50–100 μmol/L per day following complete loss of renal function in a previously normal patient. These changes in serum creatinine are not sufficiently responsive to serve as a practical indicator of glomerular filtration rate, particularly in AKI in critical care scenarios.
In the hospital situation, when AKI is detected incidentally, the cause(s) of the condition, such as fluid depletion (hypovolaemia), infection or the use of nephrotoxic drugs, are often apparent on close examination of the clinical history. The development of AKI in this setting is more likely to occur in people with pre-existing CKD. People with normal baseline kidney function usually need to sustain at least two separate triggers for the development of AKI; for example, hypovolaemia will rarely cause AKI in this setting, but when hypovolaemia occurs in the presence of nephrotoxic drugs then AKI may occur. In patients with pre-existing CKD, AKI (i.e. acute on chronic renal failure) can occur in patients with one trigger. By definition, the worse the baseline kidney function, the smaller the trigger required for the development of AKI. Irrespective of the presentation of AKI, it is wise to consider the complete differential diagnosis in all people; active exclusion of post-renal AKI and immune and inflammatory AKI should be considered in all cases. In AKI without an obvious precipitating pre-or post-renal cause, there is a greater need to consider these causes. Although the majority of patients have ATN, other causes such as rapidly progressive glomerulonephritis, interstitial nephritis, multiple myeloma or urinary tract obstruction must be screened for and systematically excluded. In addition to supportive care that is generic for all causes of AKI, disease-specific treatment may also be required. The investigation of AKI is outlined in Fig. 17.3.