Acute Coronary Syndromes
KEY CONCEPTS
![]() The cause of an acute coronary syndrome (ACS) is the rupture of an atherosclerotic plaque with subsequent platelet adherence, activation, and aggregation, and the activation of the clotting cascade. Ultimately, a clot forms composed of fibrin and platelets.
The cause of an acute coronary syndrome (ACS) is the rupture of an atherosclerotic plaque with subsequent platelet adherence, activation, and aggregation, and the activation of the clotting cascade. Ultimately, a clot forms composed of fibrin and platelets.
![]() The American College of Cardiology Foundation (ACCF), American Heart Association (AHA), and Society for Cardiovascular Angiography and Interventions (SCAI) recommend strategies, or guidelines, for ACS patient care for ST-segment elevation (STE) myocardial infarction (MI) and non–ST-segment elevation (NSTE) ACS, including guidelines for patients undergoing percutaneous coronary intervention (PCI).
The American College of Cardiology Foundation (ACCF), American Heart Association (AHA), and Society for Cardiovascular Angiography and Interventions (SCAI) recommend strategies, or guidelines, for ACS patient care for ST-segment elevation (STE) myocardial infarction (MI) and non–ST-segment elevation (NSTE) ACS, including guidelines for patients undergoing percutaneous coronary intervention (PCI).
![]() Patients with ischemic chest discomfort and suspected ACS are risk-stratified based on a 12-lead electrocardiogram (ECG), past medical history, and results of the troponin and creatine kinase (CK)–myocardial band (MB) tests. The diagnosis of MI is confirmed based on the results of the CK-MB and troponin biochemical marker tests.
Patients with ischemic chest discomfort and suspected ACS are risk-stratified based on a 12-lead electrocardiogram (ECG), past medical history, and results of the troponin and creatine kinase (CK)–myocardial band (MB) tests. The diagnosis of MI is confirmed based on the results of the CK-MB and troponin biochemical marker tests.
![]() Early reperfusion therapy with primary PCI of the infarct artery is the recommended therapy for patients presenting with STE MI within 12 hours of symptom onset.
Early reperfusion therapy with primary PCI of the infarct artery is the recommended therapy for patients presenting with STE MI within 12 hours of symptom onset.
![]() The most recent PCI ACCF/AHA/SCAI clinical practice guidelines recommend coronary angiography with either PCI or coronary artery bypass graft (CABG) surgery revascularization as an early treatment (early invasive strategy) for patients with NSTE ACS at an elevated risk for death or MI, including those with a high risk score or patients with refractory angina, acute heart failure, other symptoms of cardiogenic shock, or arrhythmias.
The most recent PCI ACCF/AHA/SCAI clinical practice guidelines recommend coronary angiography with either PCI or coronary artery bypass graft (CABG) surgery revascularization as an early treatment (early invasive strategy) for patients with NSTE ACS at an elevated risk for death or MI, including those with a high risk score or patients with refractory angina, acute heart failure, other symptoms of cardiogenic shock, or arrhythmias.
![]() In addition to reperfusion therapy, other early pharmacotherapy that all patients with STE MI and without contraindications should receive within the first day of hospitalization, and preferably in the emergency department, are intranasal oxygen (if oxygen saturation is low), sublingual (SL) nitroglycerin (NTG), aspirin (ASA), a P2Y12 inhibitor (clopidogrel, prasugrel, or ticagrelor depending on reperfusion strategy), and anticoagulation with bivalirudin, unfractionated heparin (UFH), enoxaparin, (agent dependent on reperfusion strategy), or fondaparinux. A glycoprotein (GP) IIb/IIIa inhibitor should be administered if UFH is selected as the anticoagulant for patients undergoing primary PCI. A statin should be administered prior to PCI. IV β-blockers and IV NTG should be given in selected patients. Oral β-blockers should be initiated within the first day in patients without contraindications.
In addition to reperfusion therapy, other early pharmacotherapy that all patients with STE MI and without contraindications should receive within the first day of hospitalization, and preferably in the emergency department, are intranasal oxygen (if oxygen saturation is low), sublingual (SL) nitroglycerin (NTG), aspirin (ASA), a P2Y12 inhibitor (clopidogrel, prasugrel, or ticagrelor depending on reperfusion strategy), and anticoagulation with bivalirudin, unfractionated heparin (UFH), enoxaparin, (agent dependent on reperfusion strategy), or fondaparinux. A glycoprotein (GP) IIb/IIIa inhibitor should be administered if UFH is selected as the anticoagulant for patients undergoing primary PCI. A statin should be administered prior to PCI. IV β-blockers and IV NTG should be given in selected patients. Oral β-blockers should be initiated within the first day in patients without contraindications.
![]() In the absence of contraindications, all patients with NSTE ACS should be treated in the emergency department with intranasal oxygen (if oxygen saturation is low), SL NTG, ASA, and an anticoagulant (UFH, enoxaparin, fondaparinux, or bivalirudin). High-risk patients should proceed to early angiography, and may receive a GP IIb/IIIa inhibitor. A P2Y12 inhibitor (selection of agent and timing of initiation dependent on selection of an interventional approach involving PCI or CABG surgery vs. a noninterventional approach with medical management alone) should be administered to all patients. A statin should be administered prior to PCI. IV β-blockers and IV NTG should be given in selected patients. Oral β-blockers should be initiated within the first day in patients without contraindications.
In the absence of contraindications, all patients with NSTE ACS should be treated in the emergency department with intranasal oxygen (if oxygen saturation is low), SL NTG, ASA, and an anticoagulant (UFH, enoxaparin, fondaparinux, or bivalirudin). High-risk patients should proceed to early angiography, and may receive a GP IIb/IIIa inhibitor. A P2Y12 inhibitor (selection of agent and timing of initiation dependent on selection of an interventional approach involving PCI or CABG surgery vs. a noninterventional approach with medical management alone) should be administered to all patients. A statin should be administered prior to PCI. IV β-blockers and IV NTG should be given in selected patients. Oral β-blockers should be initiated within the first day in patients without contraindications.
![]() Secondary prevention guidelines from the ACCF/AHA suggest that following MI from either STE MI or NSTE ACS, all patients, in the absence of contraindications, should receive indefinite treatment with ASA, a β-blocker, a statin, and an angiotensin-converting enzyme (ACE) inhibitor for secondary prevention of death, stroke, or recurrent infarction. The goal low-density lipoprotein cholesterol is less than 100 mg/dL (2.59 mmol/L) and ideally less than 70 mg/dL (1.81 mmol/L). A P2Y12 inhibitor should be continued for at least 12 months for patients undergoing PCI and for patients with NSTE ACS receiving a medical management strategy of treatment. Clopidogrel should be continued for at least 14 days in patients with STE MI in patients not undergoing PCI. An angiotensin II receptor blocker and a mineralocorticoid receptor antagonist should be given to selected patients. For all patients with ACS, treatment and control of modifiable risk factors such as hypertension (HTN), dyslipidemia, obesity, smoking, and diabetes mellitus are essential.
Secondary prevention guidelines from the ACCF/AHA suggest that following MI from either STE MI or NSTE ACS, all patients, in the absence of contraindications, should receive indefinite treatment with ASA, a β-blocker, a statin, and an angiotensin-converting enzyme (ACE) inhibitor for secondary prevention of death, stroke, or recurrent infarction. The goal low-density lipoprotein cholesterol is less than 100 mg/dL (2.59 mmol/L) and ideally less than 70 mg/dL (1.81 mmol/L). A P2Y12 inhibitor should be continued for at least 12 months for patients undergoing PCI and for patients with NSTE ACS receiving a medical management strategy of treatment. Clopidogrel should be continued for at least 14 days in patients with STE MI in patients not undergoing PCI. An angiotensin II receptor blocker and a mineralocorticoid receptor antagonist should be given to selected patients. For all patients with ACS, treatment and control of modifiable risk factors such as hypertension (HTN), dyslipidemia, obesity, smoking, and diabetes mellitus are essential.
![]() To determine the efficacy of nonpharmacologic treatments and pharmacotherapy, monitor patients for relief of ischemic discomfort, return of ECG changes to baseline, and absence or resolution of heart failure signs and symptoms. The most common adverse effects from pharmacotherapy of ACS are bleeding and hypotension.
To determine the efficacy of nonpharmacologic treatments and pharmacotherapy, monitor patients for relief of ischemic discomfort, return of ECG changes to baseline, and absence or resolution of heart failure signs and symptoms. The most common adverse effects from pharmacotherapy of ACS are bleeding and hypotension.
INTRODUCTION
Cardiovascular disease (CVD) is the leading cause of death in the United States and one of the major causes of death worldwide. Acute coronary syndromes (ACSs), including unstable angina (UA) and myocardial infarction (MI), are a form of coronary heart disease (CHD) that comprises the most common cause of CVD death.1 ![]() The cause of an ACS is primarily the rupture of an atherosclerotic plaque with subsequent platelet adherence, activation, and aggregation, and the activation of the clotting cascade. Ultimately, a clot forms composed of fibrin and platelets.
The cause of an ACS is primarily the rupture of an atherosclerotic plaque with subsequent platelet adherence, activation, and aggregation, and the activation of the clotting cascade. Ultimately, a clot forms composed of fibrin and platelets. ![]() The American Heart Association (AHA) and the American College of Cardiology Foundation (ACCF) recommend strategies, or guidelines, for ACS patient care for ST-segment elevation (STE) and non–ST-segment elevation (NSTE) ACS. In collaboration with the Society for Cardiovascular Angiography and Interventions (SCAI), the ACCF and AHA issue joint guidelines for percutaneous coronary intervention (PCI), including PCI in the setting of ACS. These practice guidelines are based on a review of available clinical evidence, have graded recommendations based on evidence and expert opinion, and are updated periodically. These guidelines form the cornerstone for quality care of the ACS patient.2–7
The American Heart Association (AHA) and the American College of Cardiology Foundation (ACCF) recommend strategies, or guidelines, for ACS patient care for ST-segment elevation (STE) and non–ST-segment elevation (NSTE) ACS. In collaboration with the Society for Cardiovascular Angiography and Interventions (SCAI), the ACCF and AHA issue joint guidelines for percutaneous coronary intervention (PCI), including PCI in the setting of ACS. These practice guidelines are based on a review of available clinical evidence, have graded recommendations based on evidence and expert opinion, and are updated periodically. These guidelines form the cornerstone for quality care of the ACS patient.2–7
EPIDEMIOLOGY
Each year, more than 1.1 million Americans will experience an ACS, and 150,000 die of an MI.1 In the United States, more than 7 million living persons have survived an MI.1
CVDs are the leading cause of hospitalization in the United States, resulting in about 6.2 million hospital discharges per year. Each year, there are more than 1.3 million MIs, and one in six deaths is secondary to CHD.1
The proportion of patients with MI presenting with STE MI compared with those presenting with NSTE MI decreased from approximately 80% in the early 1990s to between 25% and 30% currently.1 This may be secondary to the use of the more sensitive biomarker troponin (increasing the diagnosis of MI in the NSTE ACS group), greater use of antecedent revascularization procedures, decreased reinfarction from enhanced medical therapy after an initial event, or prevention of progression of UA to MI through more effective anticoagulant and antiplatelet therapy.
According to data from the National Registry of Myocardial Infarction, in-hospital mortality has decreased by more than 20% during the last 20 years. Improvements in care that may have contributed to this mortality reduction include greater use of guideline-recommended drugs (e.g., aspirin [ASA], β-blockers, angiotensin-converting enzyme [ACE] inhibitors and angiotensin receptor blockers [ARBs], statins, clopidogrel), reductions in the door-to-needle time for administering fibrinolytics and in the corresponding first medical contact time to primary PCI, treatments for heart failure (HF), and increased use of early coronary angiography and PCI for high-risk patients with NSTE ACS.1
In patients with STE MI, in-hospital death rates are approximately 3% in patients receiving primary PCI, 7% for patients who are treated with fibrinolytics, and 16% for patients who do not receive reperfusion therapy. In patients with NSTE MI, in-hospital mortality is less than 5%. At 6 months and 1 year, mortality and reinfarction rates are similar between STE and NSTE MI.1 Other than persistent ST-segment changes and troponin, other predictors of in-hospital mortality include older age, elevated serum creatinine (SCr), tachycardia, and HF.1,2,8 One-year mortality following STE MI ranges from 7% to 18%.2
CHD is the leading cause of premature, chronic disability in the United States. The risks of CHD events, such as death, recurrent MI, and stroke, are higher for patients with established CHD and a history of MI than for patients with no known CHD. The cost of CHD is high, with estimated direct and indirect costs of more than $195 billion.1 The estimated cost of hospitalization for MI is $14,000, and the median length of hospital stay is 3.2 days.1,2 Risk-standardized hospital mortality for MI and 30-day hospital readmission rates following MI and PCI are quality performance measures and Outcome of Care Measures that are publically reportable by the Center for Medicare and Medicaid Services.6 The Patient Protection and Affordable Care Act of 2010 links quality performance measures to hospital reimbursement.
ETIOLOGY
Endothelial dysfunction, inflammation, and the formation of fatty streaks contribute to the formation of atherosclerotic coronary artery plaques, the underlying cause of coronary artery disease (CAD).7 ![]() The predominant cause of ACS in more than 90% of patients is atheromatous plaque rupture, fissuring, or erosion of an unstable atherosclerotic plaque that occludes less than 50% of the coronary lumen prior to the event, rather than a more stable 70% to 90% stenosis of the coronary artery.9 Stable stenoses are characteristic of stable angina.
The predominant cause of ACS in more than 90% of patients is atheromatous plaque rupture, fissuring, or erosion of an unstable atherosclerotic plaque that occludes less than 50% of the coronary lumen prior to the event, rather than a more stable 70% to 90% stenosis of the coronary artery.9 Stable stenoses are characteristic of stable angina.
PATHOPHYSIOLOGY
Spectrum of ACSs
ACSs include all clinical syndromes compatible with acute MI resulting from an imbalance between myocardial oxygen demand and supply. In contrast to stable angina, an ACS results primarily from diminished myocardial blood flow secondary to an occlusive or partially occlusive coronary artery thrombus. ACSs are classified according to electrocardiogram (ECG) changes into STE MI or NSTE ACS (NSTE MI and UA) (Fig. 7-1).10 An STE MI occurs when symptoms of myocardial ischemia occur in conjunction with new STE with subsequent release of biomarkers of myocardial necrosis, mainly troponins T or I, but also creatine kinase (CK)–myocardial band (MB).2 ![]() An STE MI, formerly known as Q-wave or transmural MI, typically results in an injury that transects the thickness of the myocardial wall. Following an STE MI, pathologic Q waves are frequently seen on the ECG, indicating transmural MI, whereas such an ECG manifestation is seen less commonly in patients with NSTE MI.3 NSTE MI, formerly known as non–Q-wave or nontransmural MI, is limited to the subendocardial myocardium. Patients in this case do not usually develop a pathologic Q wave on the ECG. Moreover, an NSTE MI is smaller and not as extensive as an STE MI. NSTE MI differs from UA in that ischemia is severe enough to produce myocardial necrosis resulting in the release of a detectable amount of biomarkers, mainly troponins T or I, but also CK MB, from the necrotic myocytes in the bloodstream. The clinical significance of serum markers will be discussed in greater detail in later sections of this chapter.
An STE MI, formerly known as Q-wave or transmural MI, typically results in an injury that transects the thickness of the myocardial wall. Following an STE MI, pathologic Q waves are frequently seen on the ECG, indicating transmural MI, whereas such an ECG manifestation is seen less commonly in patients with NSTE MI.3 NSTE MI, formerly known as non–Q-wave or nontransmural MI, is limited to the subendocardial myocardium. Patients in this case do not usually develop a pathologic Q wave on the ECG. Moreover, an NSTE MI is smaller and not as extensive as an STE MI. NSTE MI differs from UA in that ischemia is severe enough to produce myocardial necrosis resulting in the release of a detectable amount of biomarkers, mainly troponins T or I, but also CK MB, from the necrotic myocytes in the bloodstream. The clinical significance of serum markers will be discussed in greater detail in later sections of this chapter.
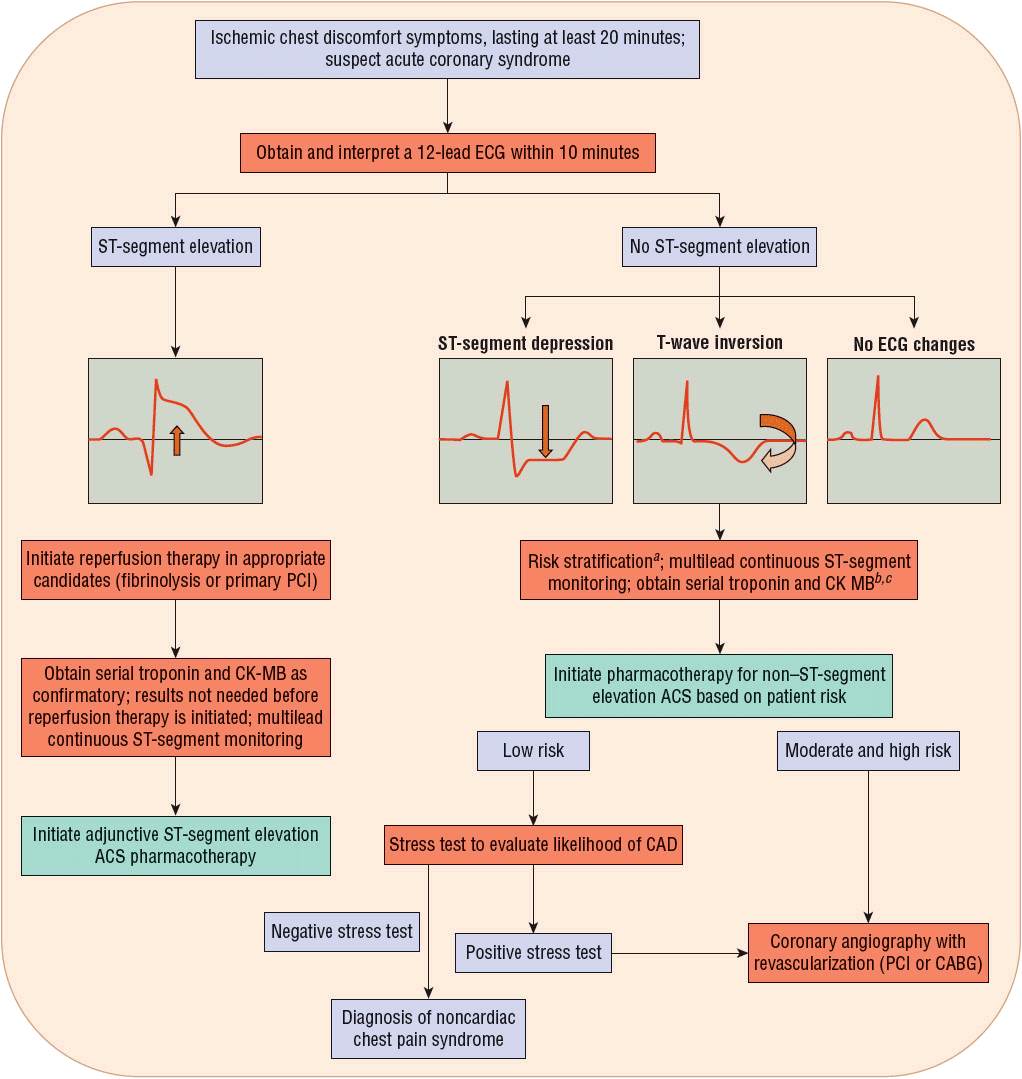
FIGURE 7-1 Evaluation of the acute coronary syndrome patient. aAs described in Table 7-1. b“Positive”: Above the myocardial infarction decision limit. c“Negative”: Below the myocardial infarction decision limit. (ACS, acute coronary syndrome; CABG, coronary artery bypass graft; CAD, coronary artery disease; CK-MB, creatine kinase myocardial band; ECG, electrocardiogram; PCI, percutaneous coronary intervention.) (Modified with permission from Spinler SA. Evolution of antithrombotic therapy used in acute coronary syndromes. In: Richardson MM, Chant C, Cheng JWM, et al., eds. Pharmacotherapy Self-Assessment Program. Book 1: Cardiology, 7th ed. Lenexa, KS: American College of Clinical Pharmacy, 2010.)
Plaque Rupture and Clot Formation
Following plaque rupture, a clot (a partially or completely occlusive thrombus) forms on top of the ruptured plaque. The thrombogenic contents of the plaque are exposed to blood elements. Exposure of collagen and tissue factor induces platelet adhesion and activation, which promote the release of platelet-derived vasoactive substances including adenosine diphosphate (ADP) and thromboxane A2 (TXA2).7 These produce vasoconstriction and potentiate platelet activation. Furthermore, during platelet activation, a change in the conformation in the glycoprotein (GP) IIb/IIIa surface receptors of platelets occurs that cross-links platelets to each other through fibrinogen bridges. This is considered the final common pathway of platelet aggregation. Inclusion of platelets gives the clot a white appearance. Simultaneously, the extrinsic coagulation cascade pathway is activated as a result of exposure of blood components to the thrombogenic lipid core and disrupted endothelium, which are rich in tissue factor. This leads to the production of thrombin (factor IIa), which converts fibrinogen to fibrin through enzymatic activity. Fibrin stabilizes the clot and traps red blood cells, which gives the clot a red appearance. Therefore, the clot is composed of cross-linked platelets and fibrin strands.9,11
Ventricular Remodeling Following an Acute MI
Ventricular remodeling is a process that occurs in several cardiovascular (CV) conditions including HF and following an MI. It is characterized by left ventricular dilation and reduced pumping function of the left ventricle, leading to cardiac failure.12 Because HF represents one of the principal causes of mortality and morbidity following an MI, preventing ventricular remodeling is an important therapeutic goal.
ACE inhibitors, ARBs, β-blockers, and mineralocorticoid receptor antagonists (MRAs) are all agents that slow down or reverse ventricular remodeling through inhibition of the renin–angiotensin–aldosterone system and/or through improvement in hemodynamics (decreasing preload or afterload).12 These agents also improve survival and will be discussed in more detail in subsequent sections of this chapter.
Complications
This chapter focuses on management of the uncomplicated ACS patient. However, it is important for clinicians to recognize complications of MI, because MI is associated with increased mortality. The most serious complication of MI is cardiogenic shock, occurring in approximately 5% to 6% of hospitalized patients presenting with STE MI.13 Mortality complicated by cardiogenic shock with MI is high, approaching 60%.14 Other complications that may result from MI are HF, valvular dysfunction, bradycardia, heart block, pericarditis, stroke secondary to left ventricular thrombus embolization, venous thromboembolism, left ventricular free wall or ventricular septal rupture, left ventricular aneurysm formation, and ventricular and atrial tachyarrhythmias.2 In fact, more than one quarter of MI patients die, presumably from ventricular fibrillation, prior to reaching the hospital.1
CLINICAL PRESENTATION Diagnosis of ACSs
Symptoms and Physical Examination Findings
The classic symptom of an ACS is midline anterior anginal chest discomfort, most often occurring when an individual is at rest, as a severe new onset, or as an increasing angina that is at least 20 minutes in duration. The chest discomfort may radiate to the shoulder, down the left arm, and to the back or to the jaw. Associated symptoms that may accompany the chest discomfort include nausea, vomiting, diaphoresis, or shortness of breath. Although similar to stable angina, the duration may be longer and the intensity greater. All healthcare professionals should review these warning symptoms with patients at high risk for CHD. On physical examination, no specific features are indicative of ACS.
Twelve-Lead ECG
There are key features of a 12-lead ECG that identify and risk-stratify a patient with an ACS. Within 10 minutes of presentation to an emergency department with symptoms of ischemic chest discomfort, a 12-lead ECG should be obtained and interpreted. When possible, a 12-lead ECG should be performed by emergency medical system providers in order to reduce the delay until myocardial reperfusion. If available, a prior 12-lead ECG should be reviewed to identify whether or not the findings on the current ECG are new or old, with new findings being more indicative of an ACS. Key findings on review of a 12-lead ECG that indicate myocardial ischemia or infarction are STE, ST-segment depression, and T-wave inversion (Fig. 7-1).10 ST-segment and/or T-wave changes in certain groupings of leads help to identify the location of the coronary artery that is the cause of the ischemia or infarction. In addition, the appearance of a new left bundle-branch block accompanied by chest discomfort is highly specific for acute MI. About one half of patients diagnosed with MI present with STE on their ECG, with the remainder having ST-segment depression, T-wave inversion, or, in some instances, no ECG changes. Some parts of the heart are more “electrically silent” than others, and myocardial ischemia may not be detected on a surface ECG. Therefore, it is important to review findings from the ECG in conjunction with biochemical markers of myocardial necrosis, such as troponin I or T, and other risk factors for CHD to determine the patient’s risk for experiencing a new MI or having other complications.
Biochemical Markers/Cardiac Enzymes
Biochemical markers of myocardial cell death are important for confirming the diagnosis of MI. The diagnosis of acute MI is confirmed when the following conditions are met in a clinical setting consistent with myocardial ischemia: “Detection of a rise and/or fall of cardiac biomarkers (cardiac troponin preferred) with at least one value above the 99th percentile of the upper reference limit with at least one of the following: (a) symptoms of ischemia; (b) new or presumed new significant ST-segment–T wave changes or new left bundle-branch block; (c) development of pathologic Q waves; or (d) imaging evidence of new loss of viable myocardium or new regional wall motion abnormality.”15 Typically, a blood sample is obtained once in the emergency department, and then 6 to 9 hours later, and in patients at a high suspicion of MI but in whom previous measurements did not reveal elevations in biomarkers, 12 to 24 hours after. A single measurement of a biochemical marker is not adequate to exclude a diagnosis of MI, as up to 15% of values that were initially below the level of detection (a “negative” test) rise to the level of detection (a “positive” test) in subsequent hours. While troponins and CK-MB appear in the blood within 6 hours of infarction, troponins stay elevated for up to 10 days while CK-MB returns to normal values within 48 hours. Hence, traditionally, CK-MB was used to detect reinfarction. However, more recent data have suggested that troponins provide similar information to CK-MB in such a situation that has led to the use of troponins in this setting as well. Current guidelines suggest that, in patients in whom a recurrent MI is suspected, a cardiac biomarker should be immediately measured, followed by a second measurement 3 to 6 hours later. A recurrent MI is diagnosed when there is an increase of at least 20% in the second measurement of the biomarker, if this value exceeds the 99th percentile of the upper reference limit.15
Risk Stratification
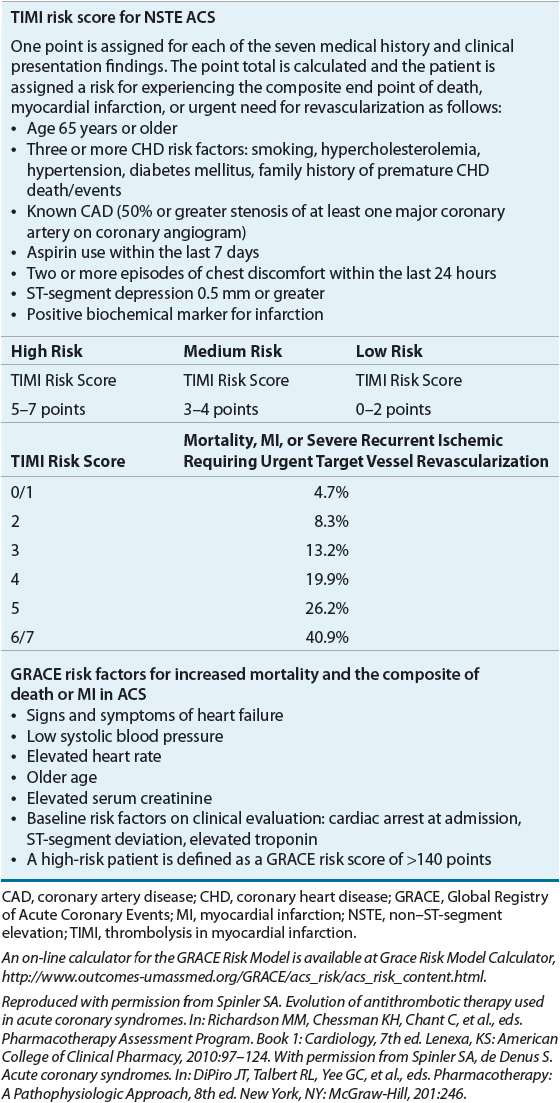
Patient symptoms, past medical history, ECG, and biomarkers, particularly cardiac troponins, are utilized to stratify patients into low, medium, or high risk of death, MI, or likelihood of failing pharmacotherapy and needing urgent coronary angiography and PCI (Table 7-1).3,5,16 ![]() Initial treatment according to risk stratification is depicted in Figure 7-1.2–5,8,10 Patients with STE MI are at the highest risk of death. Initial treatment of STE MI should proceed without evaluation of the troponins because these patients have a greater than 97% chance of having an MI subsequently diagnosed with biochemical markers. The ACCF/AHA define a target time to initiate reperfusion treatment as within 30 minutes of hospital presentation for fibrinolytics (e.g., streptokinase, alteplase, reteplase, and tenecteplase) and within 90 minutes or less from first medical contact for primary PCI.2,5 The sooner the infarct-related coronary artery is opened for these patients, the lower their mortality and the greater the amount of myocardium that is preserved.17,18 Although all patients should be evaluated for reperfusion therapy, not all patients may be eligible. Indications and contraindications for fibrinolytic therapy are described in Treatment below. Less than 25% of hospitals in the United States are equipped to perform primary PCI. If patients are not eligible for reperfusion therapy, additional pharmacotherapy for STE patients should be initiated in the emergency department and the patient transferred to a coronary intensive care unit. The typical length of stay for a patient with uncomplicated STE MI is less than 4 days.19
Initial treatment according to risk stratification is depicted in Figure 7-1.2–5,8,10 Patients with STE MI are at the highest risk of death. Initial treatment of STE MI should proceed without evaluation of the troponins because these patients have a greater than 97% chance of having an MI subsequently diagnosed with biochemical markers. The ACCF/AHA define a target time to initiate reperfusion treatment as within 30 minutes of hospital presentation for fibrinolytics (e.g., streptokinase, alteplase, reteplase, and tenecteplase) and within 90 minutes or less from first medical contact for primary PCI.2,5 The sooner the infarct-related coronary artery is opened for these patients, the lower their mortality and the greater the amount of myocardium that is preserved.17,18 Although all patients should be evaluated for reperfusion therapy, not all patients may be eligible. Indications and contraindications for fibrinolytic therapy are described in Treatment below. Less than 25% of hospitals in the United States are equipped to perform primary PCI. If patients are not eligible for reperfusion therapy, additional pharmacotherapy for STE patients should be initiated in the emergency department and the patient transferred to a coronary intensive care unit. The typical length of stay for a patient with uncomplicated STE MI is less than 4 days.19
Risk stratification of the patient with NSTE ACS is more complex because in-hospital outcomes for this group of patients vary with reported rates of death of 0% to 12%, reinfarction rates of 0% to 3%, and recurrent severe ischemia rates of 5% to 20%.17 Not all patients presenting with suspected NSTE ACS will even have CAD. Some will eventually be diagnosed with nonischemic chest discomfort. In general, among NSTE patients, those with ST-segment depression (Fig. 7-1) and/or elevated biomarkers are at higher risk of death or recurrent infarction.
TREATMENT
Desired Outcomes
Short-term desired outcomes in a patient with ACS are: (a) early restoration of blood flow to the infarct-related artery to prevent infarct expansion (in the case of MI) or prevent complete occlusion and MI (in UA); (b) prevention of death and other MI complications; (c) prevention of coronary artery reocclusion; and as evidence of restoration of coronary artery blood flow; (d) relief of ischemic chest discomfort; and (e) resolution of ST-segment and T-wave changes on the ECG.
Long-term desired outcomes are control of CV risk factors, prevention of additional CV events, including reinfarction, stroke, and HF, and improvement in quality of life.
General Approach to Treatment
Selecting evidence-based therapies described in the ACCF/AHA guidelines for patients without contraindications results in lower mortality.20–22 General treatment measures for all STE MI and high- and intermediate-risk NSTE ACS patients include admission to hospital, oxygen administration (if oxygen saturation is low, less than 90%), continuous multilead ST-segment monitoring for arrhythmias and ischemia, frequent measurement of vital signs, bedrest for 12 hours in hemodynamically stable patients, avoidance of the Valsalva maneuver (prescribe stool softeners routinely), and pain relief (Figs. 7-2 and 7-3).2–4,8,10,16
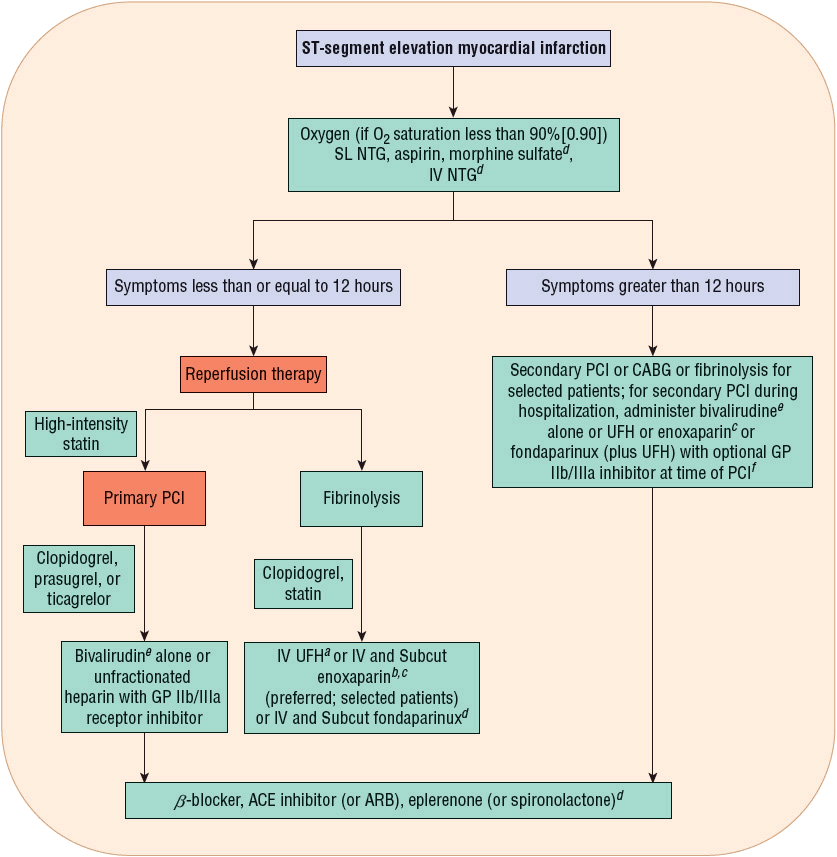
FIGURE 7-2 Initial pharmacotherapy for ST-segment elevation myocardial infarction. aFor at least 48 hours. bSee Table 7-2 for dosing and specific types of patients who should not receive enoxaparin.c For the duration of hospitalization, up to 8 days. dFor selected patients, see Table 7-2. eIf pretreated with UFH, stop UFH infusion for 30 minutes prior to administration of bivalirudin (bolus plus infusion). fIncreased risk of major bleeding and intracranial hemorrhage if a GP IIb/IIIa inhibitor is added to an anticoagulant for PCI following fibrinolysis, especially in the elderly; weight risk versus benefit. (ACE, angiotensin-converting enzyme; ARB, angiotensin receptor blocker; CABG, coronary artery bypass graft; GP, glycoprotein; NTG, nitroglycerin; PCI, percutaneous coronary intervention; Subcut, subcutaneous; SL, sublingual; UFH, unfractionated heparin.) (Modified with permission from Spinler SA. Evolution of antithrombotic therapy used in acute coronary syndromes. In: Richardson MM, Chant C, Cheng JWM, et al., eds. Pharmacotherapy Self-Assessment Program. Book 1: Cardiology, 7th ed. Lenexa, KS: American College of Clinical Pharmacy, 2010.)
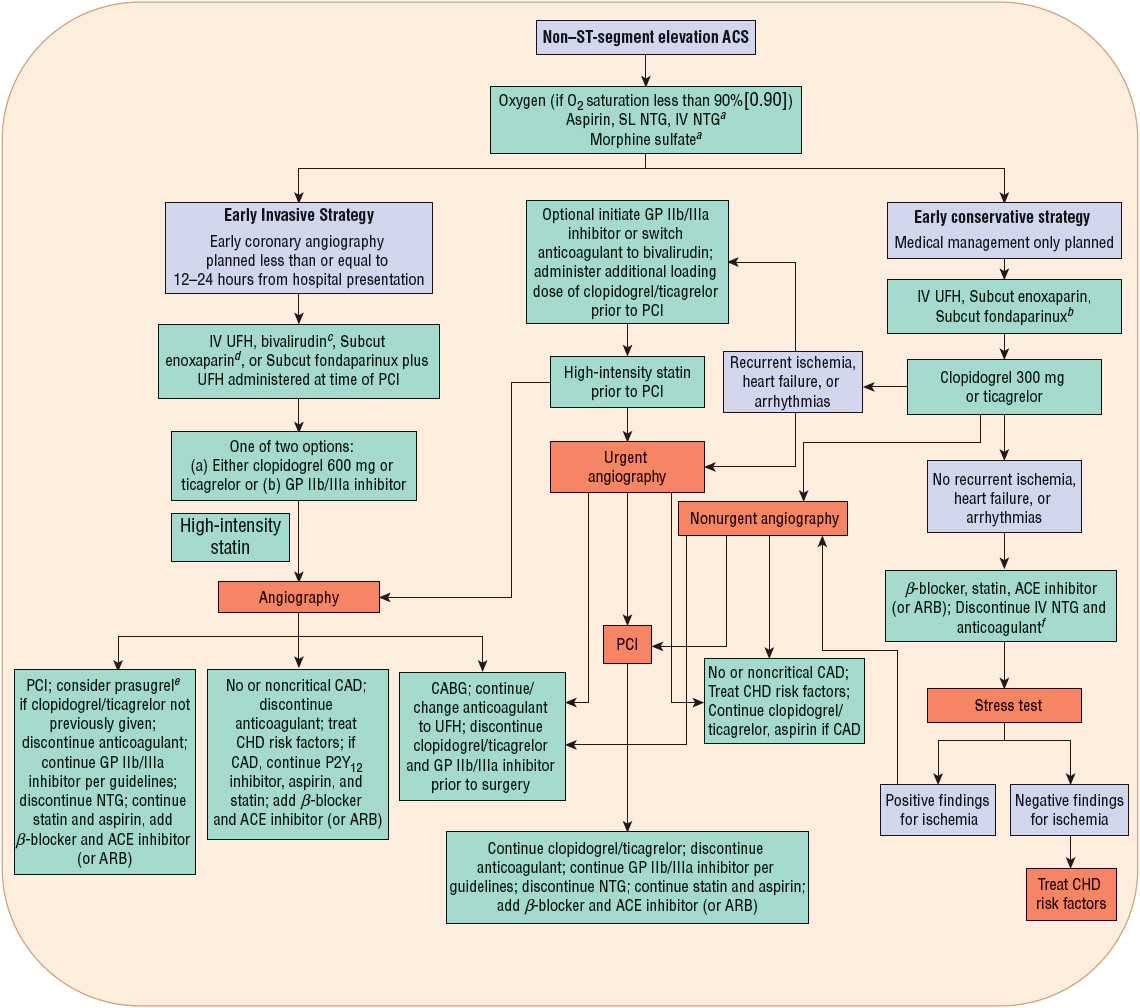
FIGURE 7-3 Initial pharmacotherapy for non–ST-segment elevation ACS. aFor selected patients, see Table 7-2. bPreferred in patients at high risk for bleeding. cIf pretreated with UFH, stop UFH infusion for 30 minutes prior to administration of bivalirudin bolus plus infusion. dMay require IV supplemental dose of enoxaparin; see Table 7-2. eDo not use if prior history of stroke/transient ischemic attack (TIA), age older than 75 years, or body weight less than or equal to 60 kg. fSubcut enoxaparin or UFH can be continued at a lower dose for venous thromboembolism prophylaxis. (ACE, angiotensin-converting enzyme; ACS, acute coronary syndrome; ARB, angiotensin receptor blocker; CABG, coronary artery bypass graft; CAD, coronary artery disease; CHD, coronary heart disease; GP, glycoprotein; NTG, nitroglycerin; PCI, percutaneous coronary intervention; Subcut, subcutaneous; SL, sublingual; UFH, unfractionated heparin.) (Modified with permission from reference 16.)
Because risk varies and resources are limited, it is important to triage and treat patients according to their risk category. Initial approaches to treatment of STE MI and NSTE ACS patients are outlined in Figures 7-2 and 7-3. Patients with STE are at high risk of death, and efforts to reestablish coronary perfusion, as well as adjunctive pharmacotherapy, should be initiated immediately.
Features identifying low-, moderate-, and high-risk NSTE ACS patients are described in Table 7-1.16,23,24
Nonpharmacologic Therapy
Primary PCI for STE MIs
Early reperfusion therapy with primary PCI of the infarct artery within 90 minutes of first medical contact is the reperfusion treatment of choice for patients presenting with STE MI who present within 12 hours of symptom onset2 ![]() (Fig. 7-2). First, emergency medical services are activated for a patient complaining of ischemic symptoms. Paramedics arrive to care for the patient out of the hospital and they perform a 12-lead ECG that demonstrates STE. Sometimes, the 12-lead ECG is transmitted electronically or via telephone to an emergency department where a physician reviews the ECG and may “activate” the cardiac catheterization medical team to alert them that a patient with STE MI will be arriving at the hospital shortly. The patient is transported to the emergency department. For primary PCI, the patient is taken from the emergency department to the cardiac catheterization laboratory and undergoes coronary angiography with either balloon angioplasty or placement of a bare metal or drug-eluting intracoronary stent in the artery associated with the infarct. In order to meet the quality of care metric of less than 90 minutes from first medical contact to primary PCI, many transitions of care occur and care coordination between paramedics, emergency department staff, and cardiac catheterization is vital. Every minute delay results in additional myocardial cell damage that may be irreversible.
(Fig. 7-2). First, emergency medical services are activated for a patient complaining of ischemic symptoms. Paramedics arrive to care for the patient out of the hospital and they perform a 12-lead ECG that demonstrates STE. Sometimes, the 12-lead ECG is transmitted electronically or via telephone to an emergency department where a physician reviews the ECG and may “activate” the cardiac catheterization medical team to alert them that a patient with STE MI will be arriving at the hospital shortly. The patient is transported to the emergency department. For primary PCI, the patient is taken from the emergency department to the cardiac catheterization laboratory and undergoes coronary angiography with either balloon angioplasty or placement of a bare metal or drug-eluting intracoronary stent in the artery associated with the infarct. In order to meet the quality of care metric of less than 90 minutes from first medical contact to primary PCI, many transitions of care occur and care coordination between paramedics, emergency department staff, and cardiac catheterization is vital. Every minute delay results in additional myocardial cell damage that may be irreversible.
About 62% of patients with STE MI are treated with primary PCI; 18% are treated with fibrinolytics. Results from a meta-analysis of trials comparing fibrinolysis with primary PCI indicate a lower mortality rate with primary PCI.25 One reason for the superiority of primary PCI compared with fibrinolysis is that more than 90% of occluded infarct-related coronary arteries are opened with primary PCI compared with fewer than 60% of coronary arteries opened with currently available fibrinolytics.2 In addition, intracranial hemorrhage (ICH) and major bleeding risks from primary PCI are lower than the risks of severe bleeding events following fibrinolysis. An invasive strategy of primary PCI is generally preferred in patients presenting to institutions with skilled interventional cardiologists and a catheterization laboratory immediately available, patients in cardiogenic shock, those with contraindications to fibrinolytics, and those with continuing symptoms 12 to 24 hours after symptom onset.2
A quality performance measure (quality performance measures are measures of quality healthcare developed from practice guidelines and intended to permit the quality of patient care to be assessed, compared between institutions and over time, and, ultimately, improved) in the care of MI patients with STE MI is the time from first medical contact to the time the occluded artery is opened with PCI. This “first medical contact-to-primary PCI” time should be equal to or less than 90 minutes.2 Efforts to improve system-wide rapid triage of patients with STE MI such as participation in the Door to Balloon Time Alliance and Get with the Guidelines (GWTG) educational programs within health systems have resulted in shorter primary PCI times in the United States. In 2010, the median door-to-primary PCI time (meaning the time from hospital arrival to primary PCI) was 64 minutes, decreasing from a median of 96 minutes in 2005.26 In 2005, 44% of patients treated with primary PCI had door-to-primary PCI times of less than 90 minutes, while in 2010 this percentage had increased to 94%.26 Unfortunately, most hospitals do not have interventional cardiology services capable of performing primary PCI 24 hours a day. Patients presenting to facilities that do not have interventional cardiology services can be transferred to such facilities when a transfer protocol that minimizes transfer delays has been established between the institutions and if primary PCI can be performed within the first 120 minutes of medical contact.2
PCI during hospitalization for STE MI may also be appropriate in other patients following STE MI, such as those in whom fibrinolysis is not successful, those presenting later in cardiogenic shock, those with life-threatening ventricular arrhythmias, and those with persistent rest ischemia or signs of ischemia on stress testing following MI.2
PCI in NSTE ACSs
The most recent PCI ACCF/AHA/SCAI clinical practice guidelines recommend coronary angiography with either PCI or coronary artery bypass graft (CABG) surgery revascularization as an early treatment (early invasive strategy) for patients with NSTE ACS at an elevated risk for death or MI, including those with a high risk score (Table 7-1) or patients with refractory angina, acute HF, other symptoms of cardiogenic shock, or arrhythmias (Fig. 7-3).5,8 ![]() Several clinical trials support an “invasive” interventional strategy with early angiography and PCI or CABG versus a “conservative medical management” strategy, whereby coronary angiography with revascularization is reserved for patients with symptoms refractory to pharmacotherapy and patients with signs of ischemia on stress testing.5,27 An early invasive approach results in a lower rate of refractory angina during hospital admission and over the first year, as well as a lower frequency of MI at 5 years, but an increased frequency of minor bleeding related to the procedure.27 An early invasive strategy is also less costly than the conservative medical stabilization approach.28 Mortality is not reduced by an early invasive approach in patients with NSTE ACS.27
Several clinical trials support an “invasive” interventional strategy with early angiography and PCI or CABG versus a “conservative medical management” strategy, whereby coronary angiography with revascularization is reserved for patients with symptoms refractory to pharmacotherapy and patients with signs of ischemia on stress testing.5,27 An early invasive approach results in a lower rate of refractory angina during hospital admission and over the first year, as well as a lower frequency of MI at 5 years, but an increased frequency of minor bleeding related to the procedure.27 An early invasive strategy is also less costly than the conservative medical stabilization approach.28 Mortality is not reduced by an early invasive approach in patients with NSTE ACS.27
All patients undergoing PCI should receive low-dose ASA therapy indefinitely. A P2Y12 inhibitor antiplatelet (clopidogrel, prasugrel, or ticagrelor) should be administered concomitantly with ASA for at least 12 months following PCI for a patient with ACS (Table 7-2).2,5 Earlier discontinuation of the P2Y12 inhibitor should be considered if the risk of bleeding outweighs the anticipated benefit in reduction in risk of death, MI, or stroke as well as stent thrombosis.2,5 A longer duration of P2Y12 inhibitor therapy may be considered for patients receiving a drug-eluting stent, as the risk of stent thrombosis is greater on cessation of dual antiplatelet therapy.2,5 Drug-eluting stents reduce the rate of smooth muscle cell growth causing stent restenosis. However, there is a delay in endothelial cell regrowth at the site of the stent that places the patient at higher risk of thrombotic events following PCI. Therefore, dual antiplatelet therapy is indicated for a longer period of time following PCI with a drug-eluting stent. Trials are ongoing evaluating the need for an extended duration in patients without ACS undergoing PCI (greater than 12 months) of P2Y12 inhibitor therapy following PCI. Regardless of whether or not a patient with NSTE ACS receives a stent, the preferred duration of P2Y12 therapy is at least a year.2,5
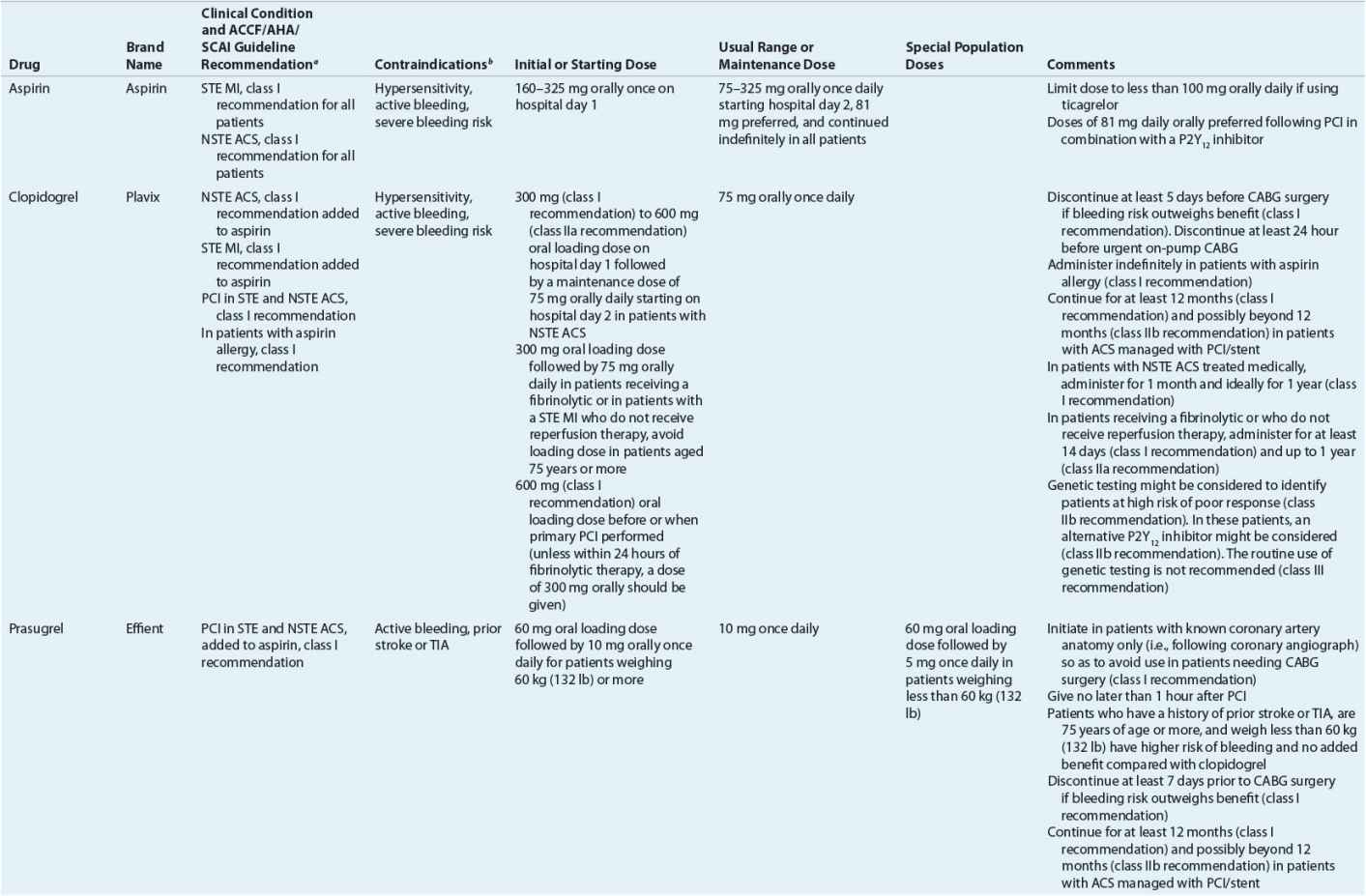
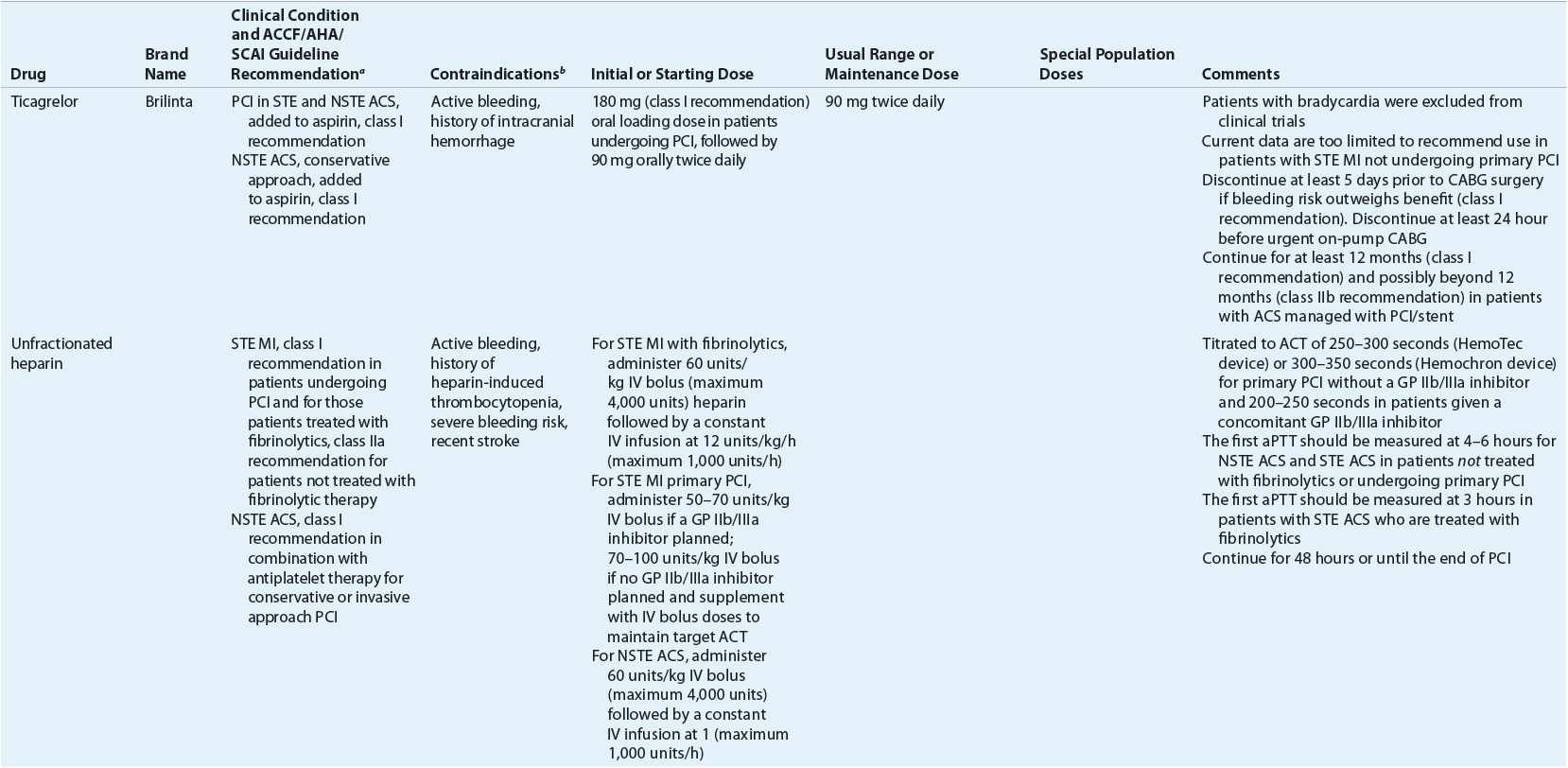
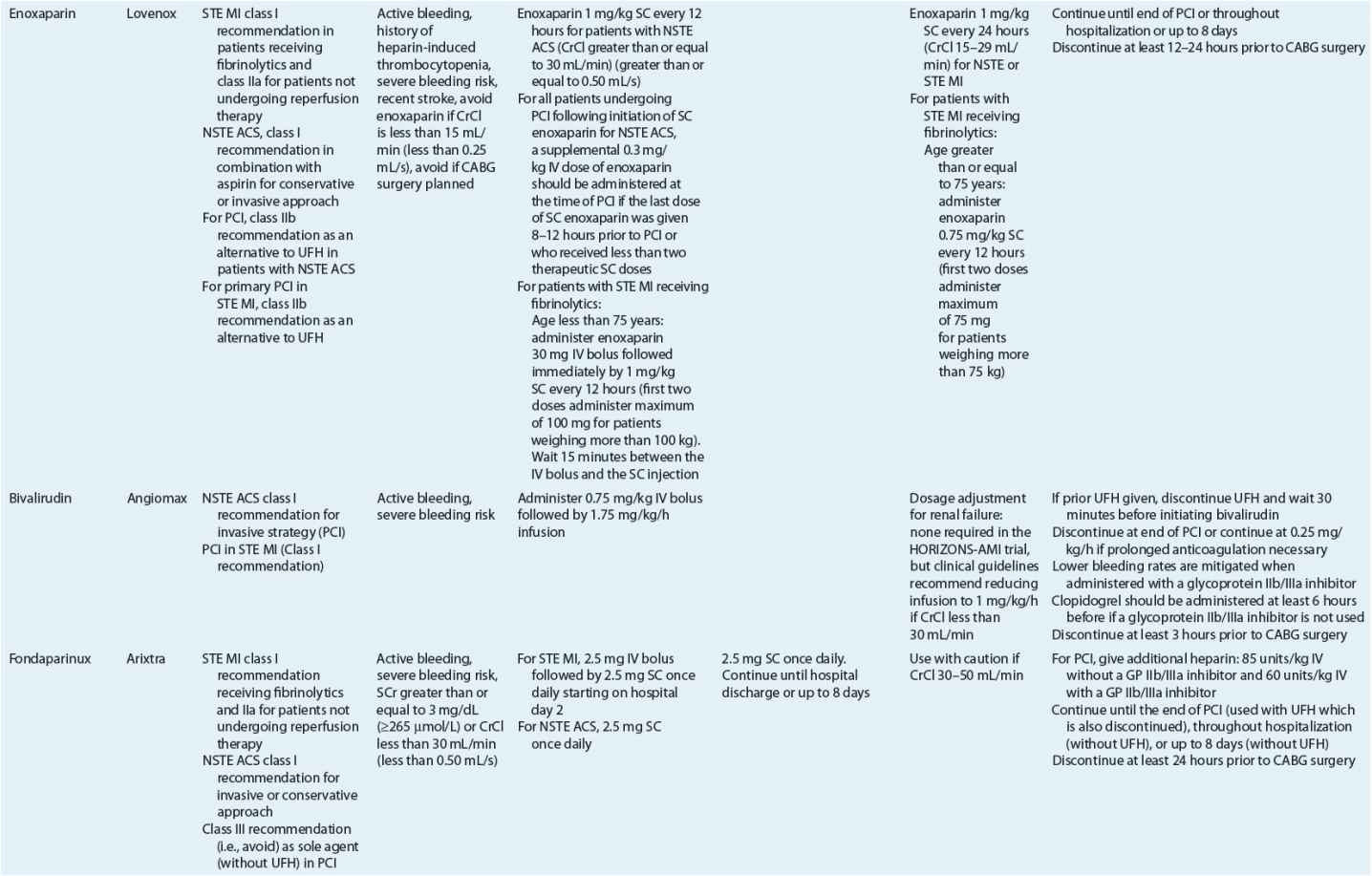
TABLE 7-2 Evidence-Based Pharmacotherapy for ST-Segment Elevation Myocardial Infarction and Non–ST-Segment Elevation Acute Coronary Syndrome2–5,8
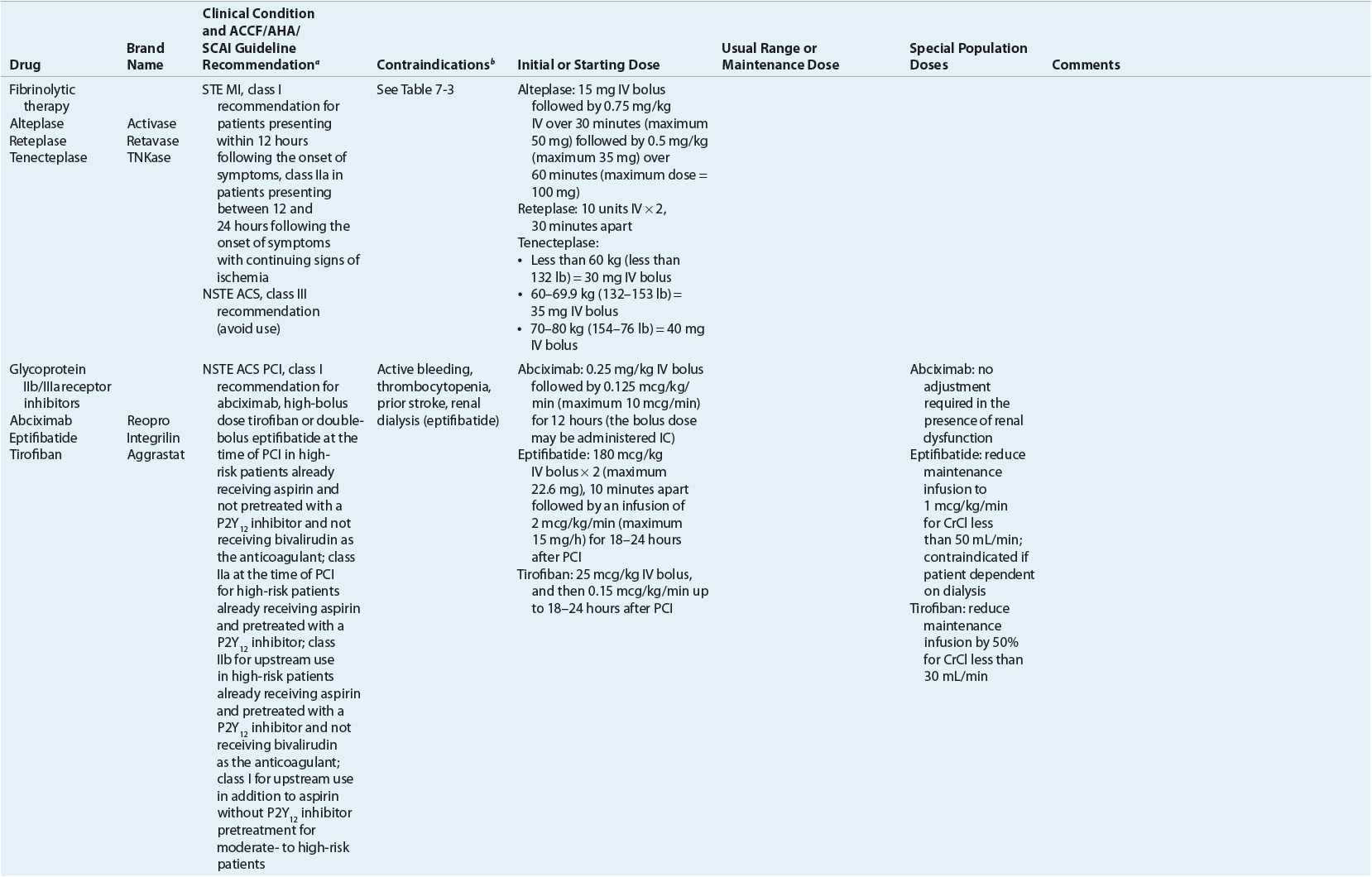
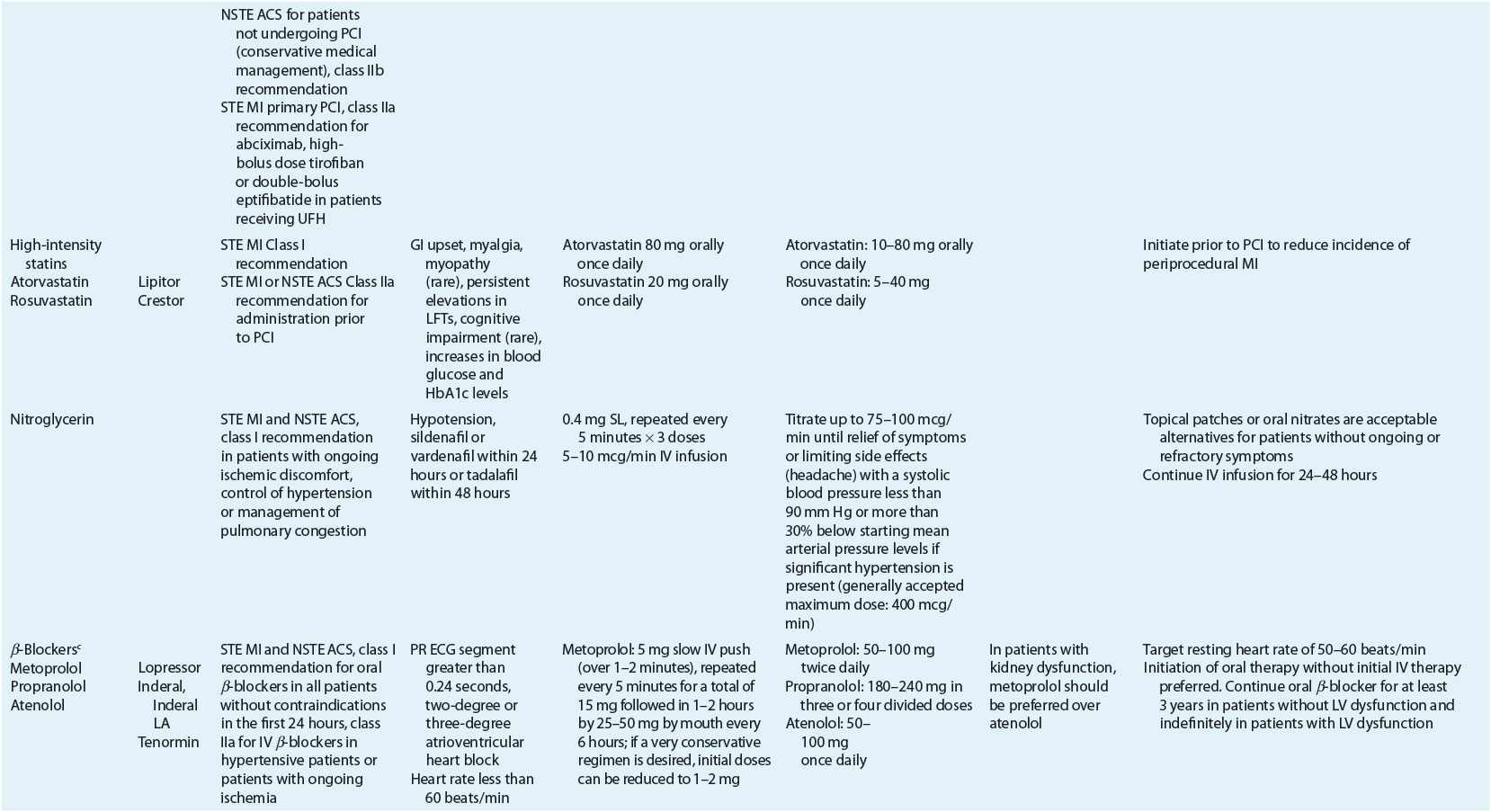
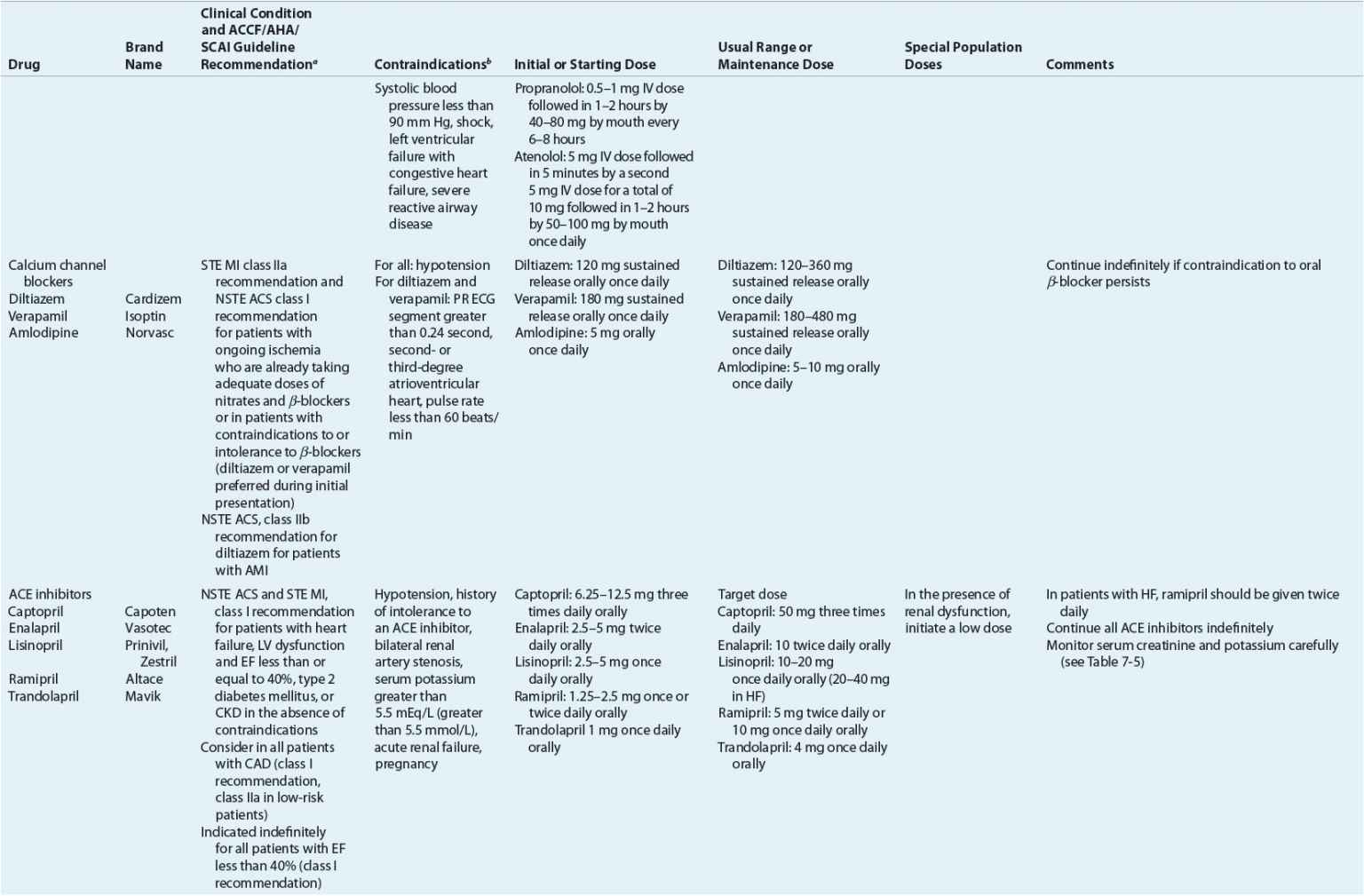
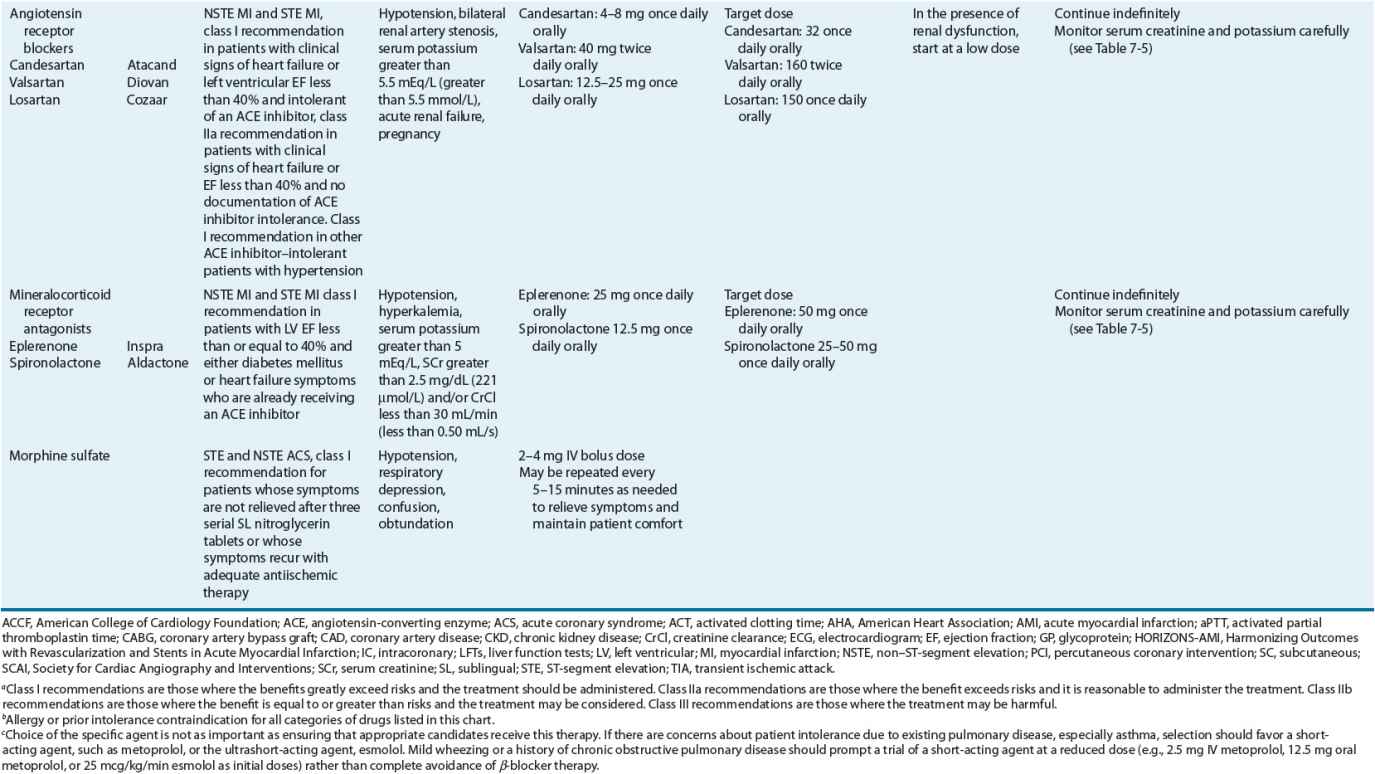
Additional Testing and Risk Stratification
For patients with NSTE ACS, an initial conservative strategy is recommended for patients with a low risk score, normal ECGs, and negative troponin tests who are without recurrence of chest discomfort (Fig. 7-3).3,4
Within the first 3 days of hospital admission patients with MI should have their left ventricular function (LVF) evaluated for risk stratification.2 The most common way LVF is measured is using an echocardiogram to calculate the patient’s left ventricular ejection fraction (LVEF). LVF is the single best predictor of mortality following MI. Patients with LVEFs less than 40% (0.40) are at highest risk of death. Patients with ventricular fibrillation or sustained ventricular tachycardia occurring more than 2 days following MI and those with LVEF less than or equal to 30% (0.30, measured at least 40 days after MI) (and have a New York Heart Association functional class I) or who have nonsustained ventricular tachycardia secondary to a prior MI and a LVEF of less than or equal to 40% (0.40) with inducible ventricular fibrillation or ventricular tachycardia at electrophysiology study benefit from placement of an implantable cardioverter-defibrillator (ICD) for sudden cardiac death prevention.29
Predischarge from the hospital, stress testing (Fig. 7-3) is indicated in patients with NSTE ACS where an initial conservative strategy is selected and for patients with STE MI where coronary angiography was not performed and there has been no recurrent ischemia.2–4 Following the stress test, patients deemed not at low risk should undergo left heart catheterization with coronary angiography and revascularization as indicated by the results.3
Early Pharmacotherapy for STE MIs
Pharmacotherapy for early treatment of ACS is outlined in Figure 7-2 and Table 7-2.2–5,8 ![]() According to the ACCF/AHA STE MI practice guidelines, in addition to reperfusion therapy, other early pharmacotherapy that all patients with STE MI and without contraindications should receive within the first day of hospitalization, and preferably in the emergency department, are intranasal oxygen (if oxygen saturation is low), sublingual (SL) nitroglycerin (NTG), ASA, a P2Y12 inhibitor (clopidogrel, prasugrel, or ticagrelor depending on reperfusion strategy), and anticoagulation with bivalirudin, unfractionated heparin (UFH), enoxaparin, or fondaparinux (agent dependent on reperfusion strategy; see Table 7-2). A GP IIb/IIIa inhibitor should be administered with UFH for patients undergoing primary PCI. IV β-blockers and IV NTG should be given in selected patients. Oral β-blockers should be initiated within the first day in patients without cardiogenic shock.2–4,8 Morphine is administered to patients with refractory angina as an analgesic and a venodilator that lowers preload. These agents should be administered early while the patient is still in the emergency department. An ACE inhibitor is recommended to be administered within the first 24 hours in patients with STE MI who have either an anterior wall MI or an LVEF less than or equal to 40% (0.40) and no contraindications. Dosing and contraindications for SL and IV NTG, ASA, clopidogrel, β-blockers, ACE inhibitors, anticoagulants, and fibrinolytics are listed in Table 7-2.2–5,8
According to the ACCF/AHA STE MI practice guidelines, in addition to reperfusion therapy, other early pharmacotherapy that all patients with STE MI and without contraindications should receive within the first day of hospitalization, and preferably in the emergency department, are intranasal oxygen (if oxygen saturation is low), sublingual (SL) nitroglycerin (NTG), ASA, a P2Y12 inhibitor (clopidogrel, prasugrel, or ticagrelor depending on reperfusion strategy), and anticoagulation with bivalirudin, unfractionated heparin (UFH), enoxaparin, or fondaparinux (agent dependent on reperfusion strategy; see Table 7-2). A GP IIb/IIIa inhibitor should be administered with UFH for patients undergoing primary PCI. IV β-blockers and IV NTG should be given in selected patients. Oral β-blockers should be initiated within the first day in patients without cardiogenic shock.2–4,8 Morphine is administered to patients with refractory angina as an analgesic and a venodilator that lowers preload. These agents should be administered early while the patient is still in the emergency department. An ACE inhibitor is recommended to be administered within the first 24 hours in patients with STE MI who have either an anterior wall MI or an LVEF less than or equal to 40% (0.40) and no contraindications. Dosing and contraindications for SL and IV NTG, ASA, clopidogrel, β-blockers, ACE inhibitors, anticoagulants, and fibrinolytics are listed in Table 7-2.2–5,8
Fibrinolytic Therapy
Administration of a fibrinolytic agent is indicated in patients with STE MI who present to the hospital within 12 hours of the onset of chest discomfort, have at least a 1 mm STE in two or more contiguous ECG leads, and are not able to undergo primary PCI within 120 minutes of medical contact.2 The mortality benefit of fibrinolysis is highest with early administration and diminishes after 12 hours.30 The use of fibrinolytics between 12 and 24 hours after symptom onset should be limited to patients with ongoing ischemia. Fibrinolytic therapy is preferred over primary PCI where there is no cardiac catheterization laboratory or there would be a delay in “door-to-primary PCI” of more than 90 minutes (of first medical contact) within the institution or 120 minutes (of first medical contact) if the patient is transferred. Indications and contraindications for fibrinolysis are listed in Table 7-3.2 It is not necessary to obtain the results of biochemical markers before initiating fibrinolytic therapy. Because administration of fibrinolytics results in clot lysis, patients who are at high risk of major bleeding (including ICH) presenting with an absolute contraindication should not receive fibrinolytic therapy, as primary PCI is preferred. In patients who have a contraindication to fibrinolytics and PCI, or who do not have access to a facility that can perform PCIs, treatment with an anticoagulant (other than UFH) for up to 8 days can be administered.
TABLE 7-3 Indications and Contraindications to Fibrinolytic Therapy Per ACC/AHA Guidelines for Management of Patients with ST-Segment Elevation Myocardial Infarction2
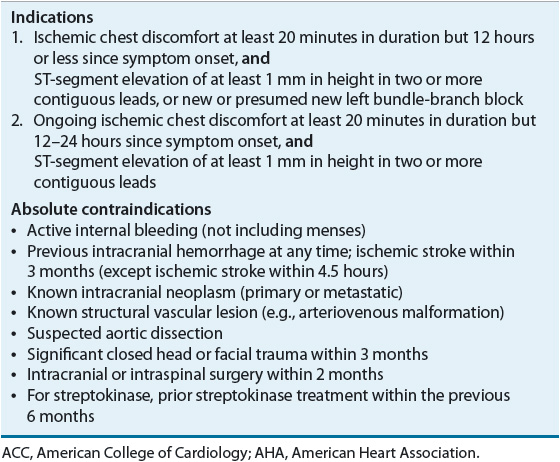
A fibrin-specific agent, such as alteplase, reteplase, or tenecteplase, is preferred over a non–fibrin-specific agent such as streptokinase.2 Fibrin-specific fibrinolytics open a greater percentage of infarcted arteries. Two trials compared alteplase with reteplase and alteplase with tenecteplase and found similar mortality between agents.31,32 Therefore, alteplase, reteplase, and tenecteplase are acceptable as first-line agents. ICH and major bleeding are the most serious side effects of fibrinolytic agents. The risk of ICH is higher with fibrin-specific agents than with streptokinase.17 However, the risk of systemic bleeding other than ICH is higher with streptokinase than with other more fibrin-specific agents and was higher with alteplase versus tenecteplase in one study.17,30–32
As mentioned previously, less than 20% of patients with STE MI receive fibrinolysis compared with more than 60% receiving primary PCI. However, 17% of eligible patients receive neither primary PCI nor fibrinolysis despite being eligible. The primary reason for lack of reperfusion therapy is that most patients present more than 12 hours after the time of symptom onset. The percentage of eligible patients who receive reperfusion therapy is a quality performance measure of care in patients with MI.33 The “door-to-needle time,” the time from hospital presentation to start of fibrinolytic therapy, is another quality performance measure.33 The ACCF/AHA guidelines recommend a “door-to-needle time” of less than 30 minutes from the time of hospital presentation until start of fibrinolytic therapy.8 The median administration time in the United States in 2006 was 29 minutes, with only 50% of patients meeting the quality performance target of less than 30 minutes.34 All hospitals should have protocols addressing fibrinolysis eligibility, dosing, and monitoring.
Aspirin
ASA is the preferred antiplatelet agent in the treatment of all ACSs.2–5,8 ASA administration to all patients who do not have contraindications to ASA therapy within 24 hours before or after hospital arrival is a quality performance measure for MI.33 The antiplatelet effects of ASA are mediated by inhibiting the synthesis of TXA2 through an irreversible inhibition of platelet cyclooxygenase-1. In patients undergoing PCI, ASA prevents acute thrombotic occlusion during the procedure. In patients receiving fibrinolytics, ASA reduces mortality, and its effects are additive to fibrinolysis alone.2,8,35 Additionally, in patients undergoing PCI, ASA, in addition to a P2Y12 inhibitor, reduces the risk of stent thrombosis.5
In patients experiencing an ACS, an initial dose equal to or greater than 160 mg nonenteric ASA is necessary to achieve a rapid platelet inhibition.36,37 Current guidelines for STE MI recommend an initial ASA dose of 162 to 325 mg (Table 7-2).2 This first dose can be chewed in order to achieve high blood concentrations and platelet inhibition rapidly. Preferably, patients undergoing PCI not previously taking ASA should receive 325 mg nonenteric-coated ASA.5 The notion of chewing ASA came from the use of an enteric-coated formulation of ASA in the Second International Study of Infarct Survival (ISIS-2) trial in order to break the enteric coating to ensure more rapid effect.35 Current data suggest that although an initial dose of 162 to 325 mg is required, long-term therapy with doses of 75 to 150 mg daily is as effective as higher doses, and therefore a daily maintenance dose of 75 to 162 mg is recommended in most patients to inhibit the 10% of the total platelet pool that is regenerated daily.37,38 In a large (n = 25,086) randomized trial, high-dose ASA, 300 to 325 mg daily, had similar frequency of CV death, MI, or stroke as well as major bleeding compared with low-dose ASA in the first 30 days following ACS presentation.39,40 Minor bleeding and GI bleeding were less frequent with low-dose ASA. In this trial, patients undergoing PCI during hospitalization had a lower frequency of death, MI, or stroke, but major bleeding was increased with high-dose ASA.43 Post hoc analysis from the Harmonizing Outcomes with Revascularization and Stents in Acute Myocardial Infarction (HORIZONS-AMI) trial compared outcomes in patients treated with ASA doses of less than or equal to 200 mg with doses of greater than 200 mg daily and found that higher doses were a predictor of major bleeding but demonstrated similar 3-year risk of CV events.41 In the Study of Platelet Inhibition and Patient Outcomes (PLATO), a randomized, double-blind clinical trial comparing ticagrelor with clopidogrel in patients receiving ASA, a post hoc analysis suggested that maintenance doses of ASA above 100 mg daily reduced the effectiveness of ticagrelor.42 Because of increased bleeding risk in patients receiving ASA plus a P2Y12 inhibitor compared with ASA alone, low-dose ASA (81 mg daily) is preferred following PCI.5,43 Low-dose ASA should be continued indefinitely.43
Nonsteroidal antiinflammatory agents other than ASA, as well as cyclooxygenase-2 (COX-2) selective antiinflammatory agents, should be discontinued at the time of STE MI secondary to increased risk of death, reinfarction, HF, and myocardial rupture.8
The most frequent side effects of ASA are dyspepsia and nausea. Patients should be counseled about the risk of bleeding, especially GI bleeding, with ASA.
Platelet P2Y12 Inhibitors
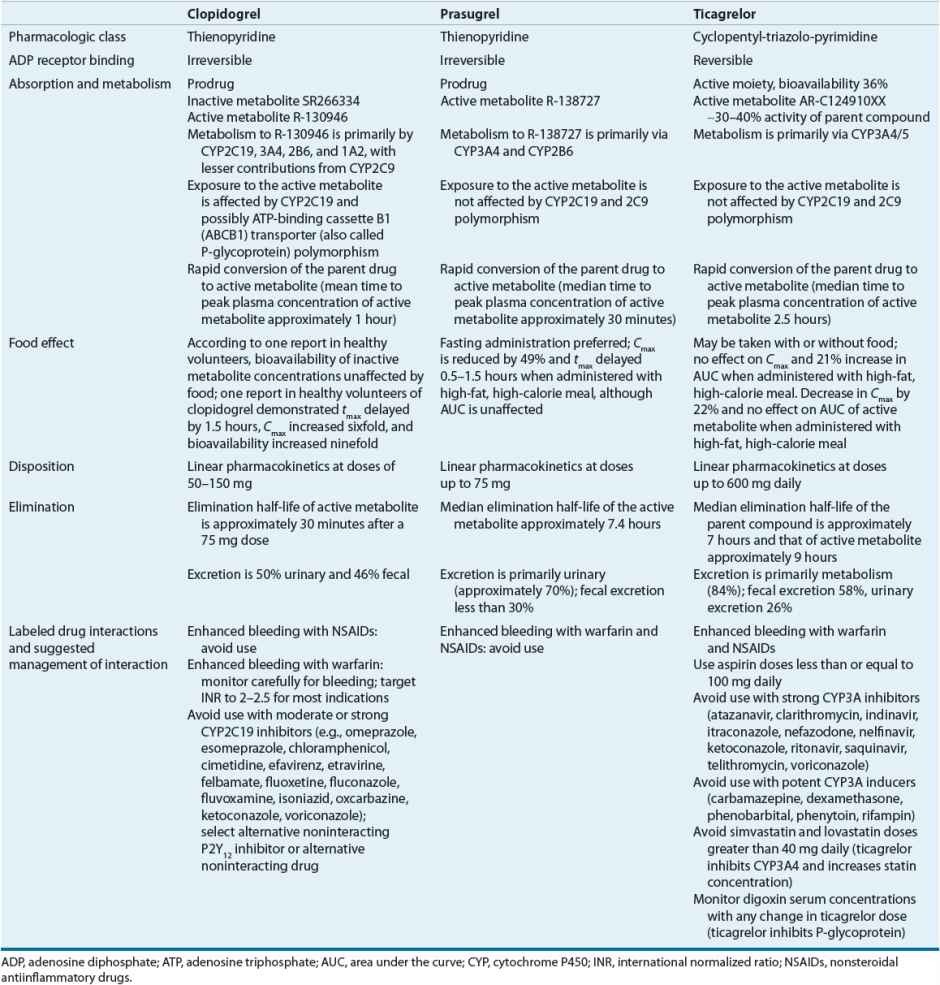
Clopidogrel, prasugrel, and ticagrelor block a subtype of ADP receptor, the P2Y12 receptor, on platelets, preventing the binding of ADP to the receptor and subsequent expression of platelet GP IIb/IIIa receptors, reducing platelet activation and aggregation. Both clopidogrel and prasugrel are thienopyridines and prodrugs that are converted to an active metabolite by a variety of cytochrome P450 (CYP) isoenzymes (Table 7-4).44–46
< div class='tao-gold-member'>