CHAPTER 10 Acquired hemolytic anemia
Introduction
Accelerated red blood cell (RBC) destruction is called hemolysis. When bone marrow compensation is adequate, hemoglobin levels remain unchanged; however, if RBC destruction surpasses production, anemia will result. Hemolytic anemia is traditionally categorized as either congenital or acquired. The term ‘acquired hemolytic anemia’ was first coined in the early 1900s1 and it is now commonly used to describe hemolytic anemia caused by antibodies (with or without complement), drugs or mechanical trauma to RBCs. Acquired hemolytic anemia can be classified as immune (autoimmune, alloimmune or drug-induced) and non-immune (infection-induced, mechanical trauma and paroxysmal nocturnal hemoglobinuria) and different causes of hemolytic anemia can overlap; for example, drug-induced hemolysis may be caused by immune mechanisms or by direct damage to the RBC membrane. In this chapter, the pathogenesis, clinical presentation and treatment of acquired hemolytic anemia will be reviewed.
Clinical and laboratory features of hemolytic anemia
The laboratory tests useful for the diagnosis of acquired hemolytic anemia include peripheral blood film examination, reticulocyte count, direct antiglobulin test (Coombs’ test), lactate dehydrogenase (LDH), bilirubin, aspartate aminotransferase (AST), haptoglobin, hemoglobinemia, methemalbumin and hemopexin, hemoglobinuria and hemosiderinuria. Bone marrow examination may be useful to uncover an underlying cause. The determination of RBC life span with radioactive isotope-labeled RBCs is rarely indicated. Laboratory tests can help determine whether the hemolytic anemia is occurring predominantly in the intravascular or extravascular space (Table 10.1). IgG-mediated autoimmune hemolysis is generally extravascular because the RBCs are destroyed by tissue macrophages in the spleen and liver. Hemolysis caused by malaria, major ABO blood group incompatibility, mechanical trauma to RBCs, thrombotic thrombocytopenic purpura (TTP) and paroxysmal nocturnal hemoglobinuria (PNH) is intravascular because RBC destruction occurs within the blood vessel.
Table 10.1 Laboratory features of intravascular and extravascular hemolysis
| Intravascular | Extravascular | |
|---|---|---|
| Serum bilirubin | Elevated | Elevated |
| Serum lactate dehydrogenase | Elevated | Elevated |
| Serum haptoglobin | Reduced or absent | Reduced or absent |
| Hemoglobinemia | Present | Absent (may be present with severe extravascular hemolysis) |
| Hemoglobinuria | Present | Absent |
| Urine hemosiderin | Present | Absent |
| Hemopexin-heme | Present | Absent (may be present with severe extravascular hemolysis) |
| Methemalbumin | Present | Absent |
| Morphology | Schistocytes, spherocytes, agglutination | Spherocytes, bite cells, blister cells, dense fragments, elliptocytes, ovalocytes, normal |
Mechanisms of hemolysis
The principal mechanisms of hemolysis include:
FcR-mediated red cell clearance
Immunoglobulins and C3b on the RBC surface target these cells for destruction by macrophages in the spleen and less frequently by Kupffer cells in the liver. These phagocytic cells express Fcγ receptors (FcγR) on their surface which bind to the Fc portion of IgG antibodies causing IgG-coated RBCs to be internalized and destroyed. Fcγ receptors are divided into three types depending on their structure, binding affinity and signalling ability: FcγRI (CD64), FcγRII (CD32) and FcγRIII (CD16). There are two extracellular immunoglobulin-like domains for FcγRII and FcγRIII, while FcγRI has three immunoglobulin-like domains. FcγRI mediates cytotoxic activity in vitro.2 It is a 72 kDa high affinity receptor (Ka = 108 − 109 M−1) capable of binding monomeric and multimeric IgG, preferentially IgG1 and IgG3; IgG3 is the most efficient IgG subclass in causing extravascular hemolysis in vivo.3,4 FcγRI is expressed on monocytes, macrophages, neutrophils and dendritic cells and is required for antibody-dependent cell-mediated cytotoxicity (ADCC), endocytosis and phagocytosis,5–8 the latter being an important mechanism of RBC destruction in IgG-mediated hemolytic anemia. Expression of FcγRI can be induced by interferon (IFN)-γ, tumor necrosis factor (TNF)-α or granulocyte colony-stimulating factor G-CSF.9–12 The family of FcγRI receptors is further divided into FcγRIA, B and C each with different affinities for binding Fc (FcγRIA with the highest and FcγRIC with the lowest) and encoded by unique genes.
FcγRII is an inhibitory receptor and acts as a negative regulator of B-cell and mast cell activation.13,14 FcγRII are low affinity receptors (Ka <107 M−1) with molecular weights of 40–43 kDa and specificity for IgG1 and IgG3. Similar to FcγRI, FcγRII is divided into FcγRIIA, B and C. FcγRIIA is more widely distributed on platelets and immune cells such as monocytes, macrophages and neutrophils and delivers signals required for phagocytosis, ADCC and cellular activation.15–17
FcγRIIB is expressed on B-cells, basophils, mast cells, monocytes, macrophages and dendritic cells and functions as an inhibitory FcR by down-regulating downstream activation signals.12,18,19 FcγRIIC is abundant on neutrophils.
FcγRIII receptors are highly glycosylated with molecular weights of 50–80 kDa. They bind multimeric IgG with an intermediate affinity (Ka = 2−3 × 107 M−1) and have the highest specificity for IgG1 and IgG3. The transmembrane form, FcγRIIIA, is found on monocytes, macrophages and eosinophils, and the GPI-linked FcγRIIIB is present on neutrophils and NK cells.12,20,21
A typical splenic macrophage has 30 000–40 000 FcR per cell.22 As a result of infection or immunization the number and affinity of FcR are upregulated through cytokines such as IFN-γ,23 which can worsen hemolysis if present. The density of FcR is also increased once IgG-coated RBCs become trapped in the spleen (and liver) and bind the FcR on the surface of phagocytes leading to receptor clustering. This effectively brings the cytoplasmic domains of multiple FcRs in close proximity, each of which share a common tyrosine-containing sequence motif called the immunoreceptor tyrosine-based activation motif (ITAM).24 The tyrosine residues are phosphorylated by Src tyrosine kinase which creates Src homology 2 (SH2)-binding sites required for signal transduction involving other members of the tyrosine kinase family. A series of downstream signals leads to the internalization of the target cell and release of cytokines.25–27
The amount and type of antibodies bound to the RBC surface is a major determinant of hemolysis.28 IgM may synergistically enhance IgG-mediated hemolysis, which is more severe when both IgG and IgM are present.29 In addition, IgG3 subclass is a more efficient mediator of phagocytosis than IgG1. IgA autoantibodies are rarely associated with autoimmune hemolysis.
Complement-mediated red cell destruction
The complement system is made up of a number of plasma proteins most of which are zymogens proteases that are activated only after they have been cleaved by another enzyme (or convertase).30,31 Many of the proteins in the complement system require calcium and activation of the complement system triggers a cascade that results in amplification of activating and inhibitory pathways32 (Fig. 10.1).
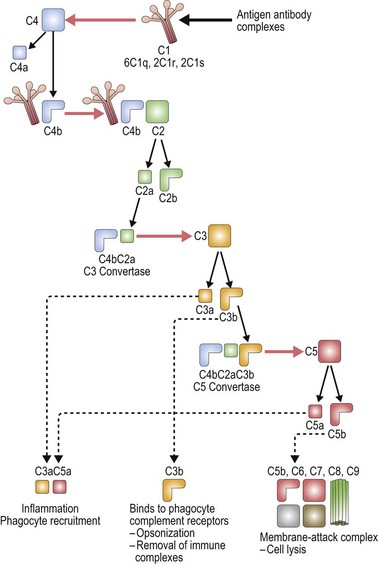
Once complement binds to RBCs, downstream events lead to the formation of the membrane attack complex and intravascular hemolysis. Complement binding is typically induced by IgM, and less frequently IgG antibodies on the surface of RBCs. Of the IgG antibodies IgG3 is most efficient at activating complement, followed by IgG1 and IgG2 (IgG4 does not). Only one bound IgM molecule is required to initiate complement activation, whereas multiple molecules of bound IgG are required. IgG dimers must be tethered within 30–40 nm (300 ångströms) of each other. The early breakdown products of complement, C3b and iC3b, are recognized by receptors on macrophages which act to contain complement activation; however, once the complement cascade proceeds to C3dg formation, the capacity for downregulation is exceeded.30
The classical pathway of complement activation begins with binding of C1q to the Fc portion of immunoglobulin on the surface of the targeted cell, and ends with the formation of the membrane attack complex. C1q is a component of the calcium-dependent C1 complex along with two molecules of C1r and two molecules of C1s. C1q has six identical subunits with globular heads and long collagen-like tails, and upon binding to Fc induces a conformational change in the C1r:C1s complex.31,33 This conformational change leads to activation of enzymatic C1r which then cleaves C1s generating an active serine protease. Activated C1s cleaves C4 producing C4b which can bind C2, anchoring it for cleavage by C1s and resulting in the production of C2a. The combined C4b2a remains covalently linked to antibody and is known as C3 convertase, the initiating factor of the classical complement pathway. C3 convertase cleaves C3; this generates C3b which covalently binds to the cell surface, and C3a which initiates a local inflammatory response. C3 is one of the most abundant proteins in plasma (1.2 mg/ml), thus providing a rich source of complement available for binding. C5 convertase (C4b2a3b) is formed by the binding of C3b to C4b2a complexes. C5 is bound to C3b and cleaved by the protease activity of C2a generating C5a and C5b.32
There are two other pathways of complement activation: the lectin pathway and the alternative pathway. The lectin pathway occurs when proteins similar to C1q, such as mannose-binding lectin, activate the complement cascade. The alternative pathway results from spontaneous hydrolysis of C3 by a distinct C3 convertase, C3bBb, and does not need a pathogen-binding protein. The classical pathway is the major pathway of complement activation in hemolytic anemia.32,34
Complement regulation
Localization of complement activation to the cell surface ensures regulation of the complement cascade.31,35 Complement activation is also controlled by inhibitor proteins including C1 inhibitor, a plasma serine protease (serpin). C1 inhibitor binds to C1r:C1s, dissociating these components from C1q, thus limiting the ability of C1s to cleave C4 and C2. Similar protective mechanisms are in place to prevent excess binding of C3 and C4. Specifically, factor I rapidly cleaves unbound C3b to iC3b (and then to C3bg), and C4b to the inactive proteins C4c and C4d. Membrane co-factor protein (MCP; CD46) acts as a co-factor for factor I by cleaving C3b and C4b, and decay-accelerating factor (DAF; CD55) also binds C3b to limit the extent of complement activation.36 DAF also promotes dissociation of the C3 convertase. Complement receptor 1 (CR1) is another membrane-bound protein that regulates complement activation by binding complement-coated particles.37 Other plasma co-factors which control C3 and C5 convertase include C4b-binding protein (C4bp) that cleaves C4b, and factor H and factor H-like protein 1 (FHL-1) that cleave C3b in the fluid phase.38 Protective mechanisms including vitronectin (S-protein) and membrane inhibitor of reactive lysis (MILR; CD59) also control the insertion of the membrane-attack complex.37,39,40
Examples of complement-mediated hemolytic anemia
IgM anti-RBC antoantibodies: Cold reactive antibodies tend to be IgM and fix C3. They often show specificity for the Ii blood group system, expressed on the ABO precursor polysaccharides.41 Binding of cold reactive autoantibodies to RBCs occurs in the peripheral circulation where temperatures are lower than the body core and may result in peripheral necrosis. Once the cells re-enter the warmer body core, the antibody dissociates but complement remains cell-bound leading to intravascular and/or extravascular hemolysis.42
Drug-induced hemolytic anemia: While most cases of drug-induced hemolysis are due to drug-dependent IgG antibodies (see later), IgM antoantibodies may also co-exist and cause intravascular hemolysis by fixing complement.42
Paroxysmal nocturnal hemoglobinuria (PNH): DAF (CD55) and MIRL (CD59) are linked to the cell surface by a phosphatidyl-inositol-glycerol (PIG) linkage. PNH is a syndrome caused by a somatic mutation in the PIG gene resulting in the functional failure of both DAF and MIRL,43 thus rendering the cells susceptible to complement attack (discussed later).
Immune hemolytic anemia
Immune hemolytic anemia is the most common form of acquired hemolytic anemia and may be autoimmune, alloimmune or drug-induced. Autoimmune hemolytic anemia (AIHA) involves the premature destruction of RBCs by autoantibodies and may be secondary to underlying disorders such as malignancy, drugs, infection and connective tissue diseases.44 RBC autoantibodies are warm or cold, based on the thermal range of activity.
Warm autoimmune hemolytic anemia (AIHA)
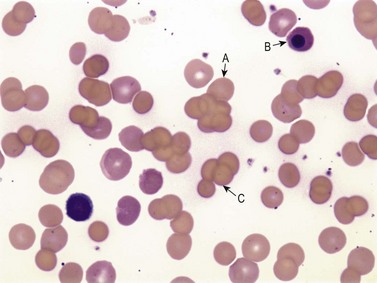
Warm AIHA accounts for more than 70% of AIHAs4 and is caused by IgG (particularly IgG1 and IgG3) directed against RBCs.28 Warm AIHA generally results in extravascular hemolysis by FcR-mediated immune clearance and is characterized by spherocytes on the blood film (Fig. 10.2); however, some degree of intravascular hemolysis may occur simultaneously because of the properties of certain IgG autoantibodies or the co-existence of IgM autoantibodies.29
Specificity of IgG autoantibodies in AIHA
In initial serologic testing, most warm autoantibodies are panagglutinins, meaning that they react with all common RBCs. These panagglutinins can be classified into different categories based on Rh specificity: (1) antibodies that react with Rh-positive cells, but not Rh-null or partially deleted RhD cells; (2) antibodies that react with a specific part of the Rh antigen system and thus show reactivity with normal RhD antigens or partially deleted RhD antigens, but not Rh-null cells; and (3) antibodies that react to all cells including Rh-null cells.45 Most warm autoantibodies (50–70%) show specificity for several antigens within the Rh antigen system;46 in one series of 150 persons with warm autoantibodies, only four were specific to a single Rh antigen (i.e. anti-e or anti-c).47 Because Rh antigens have a relatively low density on the RBC surface, autoantibodies against Rh do not cluster sufficiently to activate complement.
Up to 50% of warm autoantibodies with specificity for antigens other than Rh react with band 3 and glycophorin A.48,49 Band 3, an anion transport protein, requires a portion of glycophorin A to form the Wright(b) [Wr(b)] antigen. Di(b), an antigen of Diego system, is also carried on protein band 3 but is an uncommon target for autoantibodies.47 Decreased expression of certain blood group antigen, including Kell, Rh, MNS, Duffy, and the Kidd system50,51 may occur with the development of autoantibodies. The mechanism of this phenomenon is unknown; however, it may imply that some RBC antigens protect against hemolysis.
Some warm autoantibodies appear to have specificity against RBC antigens, yet the antibodies can still be absorbed by RBC lacking the corresponding antigen. Even the eluates made from the RBCs used for this absorption procedure demonstrate specificity for that particular antigen. The cause of this ‘pseudo-specificity’ is unclear, but has been described for the Kell, Duffy, Kidd and MNS blood group systems.50 Clinically, pseudo-specificity of the autoantibody is not directly associated with the severity of hemolysis, but it can cause confusion in the interpretation of serologic testing, especially when autoantibodies with real and pseudo-specificity co-exist.50,52 This is of practical importance when selecting compatible blood for patients with warm autoimmune hemolytic anemia.
Diagnostic evaluation of a patient with suspected AIHA
The most useful test to detect warm antibodies is the direct antiglobulin test (DAT), or Coombs’ test, using anti-IgG or anti-C3d polyclonal or monoclonal antibodies. However, the presence of antibodies and/or complement on the RBC surface does not necessarily indicate hemolysis and the strength of reactivity of the DAT is not related to clinical severity. A positive DAT without evidence of hemolysis occurs in about one per 10 000 healthy blood donors (usually IgG1). Both IgG and C3 are detected in 50% of such patients; IgG alone in 23% and C3 alone in 27%. Conversely, DAT-negative AIHA occurs in about 2% of patients with hemolysis, which may be explained by: (1) a low level of antibody sensitization; (2) hemolysis due to IgA or other immunoglobulins;53,54 or (3) variable expression of RBC antigens during the course of disease.55
The lower limit of immunoglobulin detection using the DAT is estimated to be about 100–300 antibody molecules per cell.56 Tests such as 125I-radioimmune direct antiglobulin test,57 enzyme-linked direct antiglobulin test,58,59 two-stage immunoradiometric assay with 125I-staphylococcal protein A,60 or eluate concentration may increase the sensitivity of antibody detection;4 however, the significance of these findings is uncertain.61 Before the diagnosis of Coombs’ negative autoimmune hemolytic anemia is made, other non-immune causes of hemolysis must be excluded.
In addition to detectable antibodies on the RBC surface, most patients with AIHA will have antibodies detectable in plasma (a positive indirect antiglobulin test); 57.4% of patients will have detectable antibodies on routine antibody screen, and up to 88.9% will have detectable antibody using enzyme-treated RBCs.62 If the patient requires transfusion, the challenge is to uncover co-existing alloantibodies, which may be associated with transfusion reactions, using techniques such as autoabsorption or antibody titration.
Management of patients with warm AIHA
The goal of treatment for AIHA is to suppress autoantibody production; however, the effect of immunosuppressive therapy may be delayed. Transfusion of RBCs is often required until definitive therapy is achieved. Compatible RBC units for patients with severe hemolysis and RBC antibodies may be difficult to obtain; thus, if clinically indicated, incompatible (or more precisely, the least incompatible) blood can be transfused with special care to avoid ABO and Rh incompatibility. The additional exposure to blood transfusion may further aggravate alloantibody formation;50 however, transfusion therapy is still an important supportive measure in patients whose anemia has put them at risk of serious complications.
Corticosteroids are first line therapy for AIHA,4 typically prednisone 1–1.5 mg/kg/day. The median time to response is 7–10 days. Mechanisms of steroid therapy include suppression of RBC clearance by the reticuloendothelial system,63 down-regulation of FcR,64 inhibition of release of lysosomal enzymes by macrophages and suppression of autoantibody production.65 Corticosteroids can reduce the concentration of autoantibody, but have no effect on alloantibody production.50 When hematologic improvement is seen, doses should be gradually reduced over several months to minimize complications of long-term corticosteroid use. In 60–70% of patients, complete remission can be achieved, but often maintenance therapy is required and up to 50% may relapse. If no response to corticosteroids is observed by the end of the first 3 weeks, continued therapy as sole treatment is usually ineffective. Up to 40% of patients with AIHA become dependent or resistant to corticosteroids.66,67
High-dose intravenous gamma-globulin (IVIg) results in improvement in hemolytic anemia in approximately 30–40% of patients.68–76 It is less effective than for immune thrombocytopenia (ITP) and higher doses of IVIg may be required.77 The therapeutic effect of IVIgG is usually short-lived, and further immunosuppressive therapy is often required to maintain clinical remission. The mechanism of action of IVIg therapy is likely FcR blockade,78–81 although other mechanisms such as anti-idiotype antibodies,78,82–84 interference with T-cell signaling and activation of T-suppressor cells,85–88 inhibition of B-cell maturation89–91 and dendritic-cell regulatory activity92 have been described.
Splenectomy is effective in about half of patients with AIHA,93 and like IVIg, is considerably less effective than in patients with ITP. Splenectomy removes the major site of antigen presentation and, in turn, reduces antibody production.50,94 With the advance of laparoscopic splenectomy, the incidence of severe surgical complications has been significantly reduced;95,96 however, infection, particularly with encapsulated organisms, occurs in up to 3% of patients;97 therefore, vaccination against Streptococcus pneumoniae, Neisseria meningitidis and Hemophilus influenzae type b is recommended for all patients at least 2 weeks prior to splenectomy.
Immunosuppressive therapies, including vinca alkaloids, azathioprine and cyclophosphamide, are beneficial for up to 50% of patients with AIHA,66 although the therapeutic effect may be delayed for 3–6 months and maintenance is often required. Danazol can induce long-lasting remissions in some patients;98,99 possible mechanisms include reduction in RBC-bound C3d, immunomodulation by alteration of T-cell subsets and downregulation of FcR in the reticuloendothelial (RE) system.100 Side-effects of danazol include virilization and dose-dependent hepatic toxicity.
Rituximab, a CD20 monoclonal antibody indicated for the treatment of lymphoma and rheumatoid arthritis, has been assessed in a variety of autoimmune disorders. In one report, 25 of 27 (93%) patients with primary or secondary AIHA responded to a course of rituximab after a mean of 6 weeks (range 2–16 weeks).101 Response was maintained in 18 (72%) patients after a mean of 21 months. Rituximab has been known to cause reactivation of hepatitis B102 and has been linked to the development of progressive multifocal leukoencephalopathy (PML), a rare life-threatening demyelinating disease caused by reactivation of JC polyomavirus in the brain.103
Cold autoimmune hemolytic anemia
Cold AIHA (IgM-mediated), also known as cold agglutinin disease, is much less common than warm AIHA. The antibodies react best at cold temperatures (below 30°C). In a large prospective cohort study,4 391/2390 patients (16.4%) undergoing investigations for RBC autoantibodies had cold autoantibodies while another 10 patients (0.4%) had both warm and cold autoantibodies. In another series of patients with AIHA,104 the co-existence of warm and cold autoantibodies was reported in 8% of patients. At room temperature, peripheral blood examination of patients with cold AIHA typically shows agglutination of RBCs (Fig. 10.3). Cold autoantibodies are mainly IgM (85%);4 however, the IgG biphasic Donath–Landsteiner antibody accounts for approximately 15% of such autoantibodies, especially in children.4,105
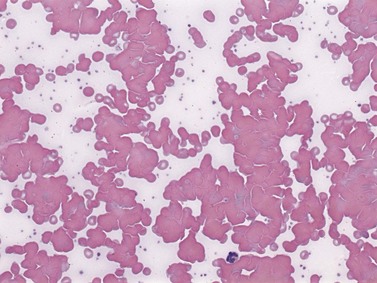
Red cell sensitization by cold IgM usually occurs in the body extremities (e.g. fingers, ears, nose) where temperatures may fall below 30°C, allowing antibody binding to occur. The complement cascade is then activated, which may result in intravascular hemolysis, giving rise to the characteristic clinical features of Raynaud’s phenomenon, acrocyanosis or gangrene; however, most patients with cold AIHA have mild symptoms or are asymptomatic. If the inhibitors of complement stop the cascade, RBCs (now coated with C3b) can be removed through extravascular phagocytosis. However, eventually C3b is degraded to C3d which does not induce clearance by reticuloendothelial tissues, but is detectable with the anti-C3d reagent used in the DAT. The most common antibody specificity is the I (‘big I’) blood group antigen, typical following Mycoplasma pneumoniae infection; anti-i (‘little i’) is often associated with infectious mononucleosis. Other less frequent specificities include P, Pr, A1, D, Vo, Gd (glycolipid dependent gangliosides),106 Lud,107 F1,108 Ju and IA.4,109 Specificity will be apparent at temperatures between 15 and 20°C. Clinically, the specificity of the antibody is less important than its thermal amplitude.
Cold avoidance is the cornerstone of management of patients with cold agglutinin disease. Corticosteroids and alkylating agents are usually ineffective, and the likelihood of remission with splenectomy is low. Plasmapheresis can be used to treat acute hemolytic episodes since it effectively removes the IgM autoantibody; this is best performed in a warm environment with prewarmed tubing and equipment. Blood transfusion, when needed, is infused at room temperature; it remains controversial whether a blood warmer offers additional benefit. If cold AIHA is secondary to an underlying neoplastic disease, chemotherapy including alkylating agents may reduce the production of cold autoantibody. Preliminary observations with rituximab are encouraging, resulting in an increase in hemoglobin concentration by 4 g/dl and a decrease in IgM levels in 54% of patients.110,111
Paroxysmal cold hemoglobinuria
Paroxysmal cold hemoglobinuria (PCH) was one of the first recognized anemias described in the mid-1800s.109 For years, PCH was believed to be a rare form of acquired AIHA associated with congenital syphilis. It then became recognized that PCH caused up to 40% of acute transient hemolytic anemia in young children112 during viral infections such as measles, mumps, chickenpox and influenza. Due to the transient nature of the disease, establishing the diagnosis is often difficult. The biphasic IgG antibody (Donath–Landsteiner antibody) is directed against globoside glycosphingolipid (P antigen) and causes hemolysis by a unique mechanism: the antibody binds to RBCs in the cooler temperatures of the peripheral circulation and activates complement causing intravascular hemolysis when RBCs return to the warmer body core. Donath–Landsteiner antibodies are more potent than IgM cold agglutinins in initiating intravascular hemolysis because they retain their binding affinity even when the RBCs return to warmer temperatures.44,113 IgG3 is the major immunoglobulin subclass for Donath–Landsteiner antibodies.114
Direct and indirect Donath–Landsteiner tests establish the diagnosis of PCH.115 The direct test is done by incubating a whole blood sample at 0°C for 1 h, and then at 37°C for an additional 30 min. If the Donath–Landsteiner antibody is present, it will bind to RBC during the cold incubation phase and lyse the cells during the warm phase. As a control, a whole blood sample maintained at 37°C should show no evidence of hemolysis. The indirect test is done by mixing the patient’s serum with ABO compatible P-positive RBCs in the presence of fresh serum as a source of complement. The indirect test has a much higher sensitivity than the direct test because of: (1) the addition of a complement source; (2) the ability to adjust the serum to cell ratio; and (3) the increased susceptibility of donor RBCs to lyse (compared with patient cells, which are coated with C3d). The sensitivity of the test can be increased further by using enzyme-treated RBCs. False positive results occur with IgM hemolytic antibodies with a high thermal range. False negative results may be seen with low antibody titers or if soluble globoside (P antigen) is present in the added fresh normal serum.105
The management of PCH may require urgent blood transfusion depending on the level of anemia and clinical symptoms. Theoretically, P antigen-negative RBCs may minimize in vivo hemolysis; however, P-negative blood is usually not available and the transfusion of P-positive RBCs can be beneficial. Transfusions should be administered slowly while the patient is kept warm.116 Neither washing RBCs117 (to remove complement proteins in donor plasma) nor corticosteroid therapy have been shown to be effective in PCH.105
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


