Absorption
OBJECTIVES
The reader will be able to:
- Define the following terms: absorption rate-limited, Biopharmaceutics Classification System (BCS), dissolution, dissolution rate-limited, first-pass loss, gastric emptying, gastrointestinal absorption, intestinal transit, saturable first-pass metabolism.
- Describe the steps involved in the systemic absorption of a drug after extravascular administration generally and specifically after oral administration.
- List five factors influencing dissolution rate of a drug.
- Distinguish between dissolution and permeability-rate limitations in systemic absorption after oral administration.
- State the mean transit times of material in the fasted stomach, small intestine, and large intestine.
- Anticipate for which compounds intestinal permeability is likely to be the reason for incomplete oral bioavailability for a drug absorbed by passive diffusion.
- List possible competing reactions responsible for low oral bioavailability of drugs, together with one example of each possibility.
- Calculate the anticipated maximum oral bioavailability of a drug given either its hepatic extraction ratio or the appropriate information to estimate this value under nonsaturating conditions.
- Anticipate the influence of physicochemical properties of a drug on its absorption from different sites of administration.
- Anticipate the role of gastric emptying and intestinal transit in the systemic absorption of a drug given orally with particular reference to the physicochemical properties of the drug and its dosage form.
- Anticipate the influence of food on the systemic absorption of a drug given orally.
- Describe the rationale underpinning the application of the BCS to the waiver of bioequivalence studies of multisource formulations of the same drug and to deciding which drug transporters are unlikely to affect either their absorption or disposition.
- Describe the influence of decreased permeability and surface area along the intestinal tract on the performance of oral constant-rate release dosage forms.
 his chapter deals with the general physiologic and physicochemical principles governing systemic drug absorption, particularly following oral administration. Being a complex structure, many anatomic and physiologic factors affect the overall rate and extent of drug absorption from the gastrointestinal tract, making precise quantitative predictions difficult. Nonetheless, some generalizations can be made on the consequences of many of the events occurring at this and other sites of absorption.
his chapter deals with the general physiologic and physicochemical principles governing systemic drug absorption, particularly following oral administration. Being a complex structure, many anatomic and physiologic factors affect the overall rate and extent of drug absorption from the gastrointestinal tract, making precise quantitative predictions difficult. Nonetheless, some generalizations can be made on the consequences of many of the events occurring at this and other sites of absorption.
Passage of drug through the membranes dividing the absorption site from the blood is a prerequisite for absorption to occur. To do so, a drug must be in solution. Most drugs are administered as solid preparations (e.g., tablets and capsules). Because solid particles cannot pass through membranes, a drug must dissolve to be absorbed. Many factors influence the dissolution, or release, of drug from a solid pharmaceutical formulation. However, before discussing the factors influencing the release, let us first consider drug absorption from a solution.
ABSORPTION FROM SOLUTION
Systemic absorption is favored after extravascular administration because the body acts as a sink, producing a concentration difference between the diffusible unbound concentrations at the absorption site and in systemic blood. The concentration gradient across the absorptive membranes is maintained by distribution to tissues and elimination of absorbed drug. Physiologic and physicochemical factors that determine movement of drug through membranes in general are presented in Chapter 4, Membranes and Distribution. Included among them are the molecular size of the drug, the nature of the membrane involved, presence of transporters, perfusion, and pH. These factors and others are now considered with respect to drug passage through gastrointestinal membranes. In this context, gastrointestinal absorption is the term that is subsequently used for this process.
GASTROINTESTINAL ABSORPTION
The first potential site for absorption of orally administered drugs is the stomach. In accordance with the prediction of the pH partition hypothesis, weak acids are absorbed more rapidly from the stomach at pH 1.0 than at pH 8.0, and the converse holds for weak bases. Absorption of acids, however, is much faster from the less acidic small intestine (pH 6.6 to 7.5) than from the stomach. These apparently conflicting observations can be reconciled. Surface area, permeability and, when perfusion rate-limits absorption, blood flow are important determinants of the rapidity of absorption. The intestine, especially the small intestine, is favored on all accounts. The total absorptive area of the small intestine, produced largely by microvilli, has been calculated to be about 200 m2, and an estimated 1 L of blood passes through the intestinal capillaries each minute. The corresponding estimates for the stomach are only 1 m2 and 150 mL/min. The permeability of the intestinal membranes to drugs is also greater than that of the stomach. These increases in surface area, permeability, and blood flow more than compensate for the decreased fraction of un-ionized acid in the intestine. Indeed, the absorption of all compounds—acids, bases, and neutral compounds—is faster from the small intestine than from the stomach. Because absorption is greater in the small intestine, the rate of gastric emptying can be a controlling step in the speed of drug absorption even when given in solution. In the following discussion, the examples chosen are either drugs administered as solutions or where there is reason to expect, based on solubility considerations for example, that the drug dissolves within the time it spends within the stomach, usually about 30 min or less prior to entering the small intestine. This is especially the case when a drug is taken on an empty stomach with a glass of water.
Gastric Emptying. Food, especially fat, slows gastric emptying, which explains why drugs are frequently recommended to be taken on an empty stomach when a rapid onset of action is desired. Drugs that influence gastric emptying also affect the speed of absorption of other drugs, as shown in Fig. 7-1 for acetaminophen, a common analgesic/antipyretic, when remifentanil (Ultiva), a potent ultra short-acting opioid, is concurrently administered. The observed slowing of systemic absorption, which is common to all opioids, is partially a result of slowed gastric emptying, which contributes to the constipation seen with this class of drugs. The figure also shows that gastric emptying may be slightly faster when lying on the right side than when lying on the left side (not statistically significant). Although on first glance it would appear that the extent of absorption (area under the curve [AUC]) is affected by remifentanil or the position of the body, one cannot be certain because of the limited time over which blood samples are taken. The total AUC (not provided) is needed. Clearly, it is unlikely that one would get a rapid onset of response to a drug when gastric emptying is so slowed.
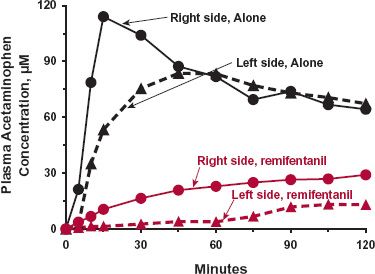
FIGURE 7-1. Gastric emptying can rate-limit systemic drug absorption. Remifentanil, an ultra short-acting opioid narcotic analgesic, slows gastric emptying as observed by the mean systemic exposure–time profiles of acetaminophen after its oral administration (1.5 g dissolved in 200 mL of water) to 10 healthy volunteers in the presence and absence of the narcotic. The drug was given alone (black lines) and 10 min after the start of a constant-rate intravenous infusion of remifentanil of 0.2 (μg/kg per minute (colored lines). Posture was also shown to minimally influence gastric emptying as observed when acetaminophen was taken while the subjects were lying on their left side with the head lower than the feet by 20 degrees (dotted lines) than when the subjects were on their right side with the head higher than the feet by 20 degrees (solid lines). (From: Walldén J, Thörn SE, Wattwil M. The delay of gastric emptying induced by remifentanil is not influenced by posture. Anesth Analg 2004;99:429–434.)
Retention of acetaminophen in the stomach increases the percentage of a dose absorbed through the gastric mucosa, but this is a minor pathway; the majority of the dose is still absorbed through the intestinal epithelium. In this regard, the stomach may be viewed as a repository organ from which pulses of drug are ejected by peristalsis onto the absorption sites in the small intestine.
Intestinal Absorption and Permeability. Throughout its length, the intestine varies in its multifaceted properties and luminal composition. The intestine may be broadly divided into the small and large intestines separated by the ileocecal valve. Surface area per unit length decreases from the duodenum to the rectum. Electrical resistance, a measure of the degree of tightness of the junctions between the epithelial cells, is much higher in the colon than in the small intestine. Proteolytic and metabolic enzymes, as well as active and facilitated transport systems are distributed variably along the intestine, often in restrictive regions. The colon abounds with anaerobic microflora. The mean pH, 6.6, in the proximal small intestine rises to 7.5 in the terminal ileum, and then falls sharply to 6.4 at the start of the cecum before finally rising again to 7.0 in the descending colon. Mean transit time of materials is around 3 to 4 hr in the small intestine and from 10 to 36 hr or even longer in the large bowel. Although these and other complexities make precise quantitative prediction of intestinal drug absorption difficult, several general features emerge.
The permeability-surface area (P · SA, see Chapter 4, Membranes and Distribution) product tends to decrease progressively from duodenum to colon. This applies to all drug molecules traversing the intestine epithelium by non–carrier-mediated processes, whether via the transcellular (through cell) or paracellular (around cell) routes, when drug solutions are placed in different parts of the intestine. How much of this decrease in absorption is a result of a decrease in permeability and how much of the decrease in surface area between small and large intestine is not known for certain. For permeable drugs, absorption is rapid and probably complete within the small intestine. Even if some drugs were to enter the large intestine, the permeability there is still sufficiently high to ensure that all that entered would be absorbed. For these drugs, although absorption across the intestinal epithelia may be perfusion rate–limited, as mentioned previously, the overall rate–limiting step in systemic absorption is likely to be gastric emptying.
Absorption of less permeable, generally more polar, drugs still primarily occurs within the small intestine rather than the large intestine. Evidence supporting this notion is provided with the H2-antagonist ranitidine (Fig. 7-2). When given intracolonically, the extent of absorption is greatly reduced compared to when placed into the stomach or jejunum, as reflected by the reduced AUC (Fig. 7-2A). Evidently, very little ranitidine is absorbed from the large intestine even though drug can be there for 24 hr or more. Furthermore, absorption from the large intestine appears to become rate limiting in the elimination of drug from the body, as reflected by an increased terminal half-life after colonic administration (Fig. 7-2B).
As discussed in Chapter 4, Membranes and Distribution, molecular size is a particularly important determinant of permeability. Small polar substances, such as the antiviral acyclovir (250 g/mol), H2-antagonist cimetidine (252 g/mol), and atenolol (266 g/mol), primarily move paracellularly across the epithelium. Permeability, in general, and paracellular permeability, in particular, appears to drop off sharply with molecular weights (MWs) above 350 g/mol, a value at which compounds appear to approach the molecular dimensions of the tight junctions between intestinal cells. Drugs with poor permeability characteristics show low oral bioavailability not only for this reason, but also because of the limited time, 2 to 4 hr, they spend in the small intestine where permeability is highest. For example, when given orally, only 60% of ranitidine is absorbed and virtually all within the first 3 to 4 hr after administration; the rest is recovered unchanged in feces. The relationship between fraction absorbed intestinally and jejunal permeability of many drugs is shown in Fig. 7-3 (page 188). Clearly, low permeability drugs are likely to be poorly absorbed. Examples that are particularly poorly absorbed drugs (F = 0.005 to 0.14) are listed in Table 7-1. The drugs in the table share the common property of being polar and, with the exception of pyridostigmine, relatively large (MW >400 g/mol). Pyridostigmine is a quaternary ammonium compound with a permanent positive change, which may explain its low oral bioavailability (F about 0.07 to 0.14) despite its relatively small size (181 g/mol).
Most of the compounds in Table 7-1 (page 188) are not reliably active systemically when given orally; they must be given parenterally. Not so much because of their low oral bioavailability, but because of their excessively variable oral absorption and sometimes because there is a need for very large doses to adequately achieve a therapeutic systemic exposure. The bisphosphonates, alendronate, and ibandronate are exceptions. They are given orally in spite of their low oral bioavailabilities (about 0.005 to 0.007). The doses given are not excessively large; only small amounts are apparently needed systemically. The amount of alendronate systemically absorbed when given in a 70-mg weekly regimen is only about 0.42 mg. The amount of ibandronate systemically absorbed from a 150-mg once-monthly dose is only 0.9 mg. Pyridostigmine is also given orally to treat myasthenia gravis, in spite of its low oral bioavailability. Although oral administration is ruled out for many of the antimicrobial agents listed in Table 7-1, occasionally they have utility in treating diseases of the alimentary canal itself. The use of vancomycin in treating pseudomembranous colitis is an example.
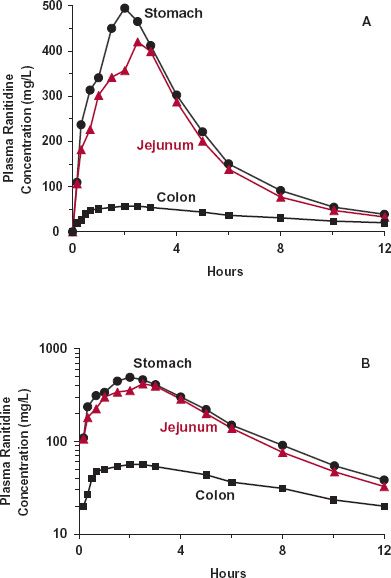
FIGURE 7-2. The gastrointestinal absorption of ranitidine varies with site of application. The variation is shown in linear (A) and semilogarithmic (B) plots of the mean plasma concentration–time profiles observed after placing an aqueous solution (6 mL) containing 150 mg of ranitidine hydrochloride into the stomach (black circles), jejunum (colored triangles), and colon (black squares) of eight volunteers, via a nasoenteric tube. The much less extensive absorption of this small (MW = 313 g/mol) polar molecule from the colon is consistent with the idea that the P · SA product is much lower in the colon than in the small intestine. Notice that absorption of ranitidine effectively ceases (in terminal decline phase) by 3 hr when placed in the stomach or jejunum, even though the drug is incompletely bioavailable (F = 0.6, data not shown), suggesting that the small intestine is the major site of absorption when ranitidine is taken orally. Also, notice in B that the terminal half-life of the decline in the plasma concentration is longer when the drug is administered into the colon. Absorption from the colon thus appears to be slow and rate limiting drug elimination. (From: Williams MF, Dukes GE, Heizer W, et al. Influence of gastrointestinal site of drug delivery on the absorption characteristics of ranitidine. Pharm Res 1992;9:1190–1194.)
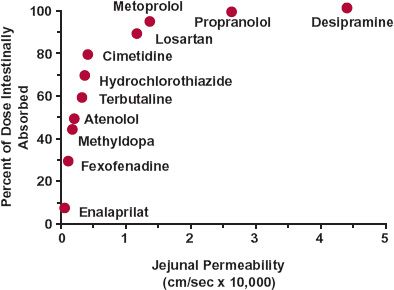
FIGURE 7-3. The fraction of a dose intestinally absorbed after oral administration correlates with human jejunal permeability. Drugs with permeabilities less than 1.0 × 10−4 cm/sec are likely to be incompletely intestinally absorbed. The lower the permeability is below this value, the greater is the likelihood of their being so. (From: Petri N, Lennernäs H. In vivo permeability studies in the gastrointestinal tract. In: van de Waterbeemd H, Lennernäs H, Artusson P, eds. Drug Bioavailability, Estimation of Solubility, Permeability, Absorption and Bioavailability. Berlin, Germany: Wiley-VCH, 2003:345–386.)

An additional determinant of permeability and hence absorption are transporters, particularly those on the apical side of the epithelial cells (i.e., facing the intestinal lumen). Uptake transporters, such as peptide transporter 1 (PepT1) and organic anion-transporting polypeptide 3 (OATP3), facilitate the absorption of amoxicillin, L-dopa, and antiepileptic/analgesic gabapentin, all of which would otherwise be poorly absorbed. Indeed, as discussed in Chapter 4, Membranes and Distribution, it is because the inherent passive permeability of such molecules is low that transporters exert an influence. In contrast, systemic absorption of some drugs is reduced by the presence of efflux transporters (e.g., P-glycoprotein). Drug examples include digoxin, used in treating heart failure and atrial fibrillation; fexofenadine, an antihistamine; paclitaxel, an anticancer drug; and saquinavir, an antiretroviral agent. Low apparent permeability results, not so much from inability to cross intestinal membranes, but from the action of the efflux transporter. Concurrent administration of inhibitors of the transport system (e.g., erythromycin, an antibiotic; ketoconazole, an antifungal agent; quinidine, used in atrial fibrillation; and ritonavir, a protease inhibitor) can increase the oral absorption of these transported substrates, whereas coadministration of inducers of this efflux transporter (e.g., rifampin, an antitubercular drug, and St. John’s wort, alleged to be useful for depression and other mental disorders) have the opposite effect. This is clearly illustrated by the effect of rifampicin on digoxin pharmacokinetics (Fig. 7-4). Digoxin, a large (781 g/mol) molecule containing three sugar groups, is absorbed and predominantly renally eliminated unchanged; bioavailability is usually about 70% and most absorption appears to occur within the first 4 hr of administration, in keeping with limited absorption beyond the small intestine. Rifampicin pretreatment has no material effect on the disposition kinetics of digoxin but, rather, reduces its oral bioavailability (based on decreased total AUC). A slowing in the speed of its absorption is also clearly indicated by the decrease in Cmax and the increase in tmax. The mechanism is an increase in the intestinal expression of P-glycoprotein induced by rifampicin. The absorption of digoxin is permeability limited; therefore, a decrease in the effective permeability of digoxin decreases both the speed and extent of its absorption.
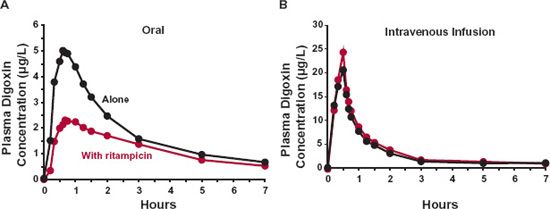
FIGURE 7-4. Rifampicin pretreatment reduces the absorption of digoxin. Shown are plots of mean plasma digoxin concentration-time profiles after oral and i.v. administration (as a 30-min infusion) of 1 mg digoxin alone ( ) and after 10 days rifampicin pretreatment (600 mg daily,
) and after 10 days rifampicin pretreatment (600 mg daily,  ) to seven healthy adults. A clear depression in the oral absorption of digoxin is inferred by the lower concentrations after oral but not i.v. administration after rifampicin pretreatment. This was corroborated by a 30 % decrease in total AUC (0–144hr) (from 54.8 to 38.2 μg-hr/L), corresponding to a fall from 63% to 38% in oral bioavailability. (From: Greiner B, Eichelbaum M, Fritz P, et al. The role of intestinal P-glycoprotein in the interaction of digoxin and rifampin. J Clin Invest 1999;104:147–153.)
) to seven healthy adults. A clear depression in the oral absorption of digoxin is inferred by the lower concentrations after oral but not i.v. administration after rifampicin pretreatment. This was corroborated by a 30 % decrease in total AUC (0–144hr) (from 54.8 to 38.2 μg-hr/L), corresponding to a fall from 63% to 38% in oral bioavailability. (From: Greiner B, Eichelbaum M, Fritz P, et al. The role of intestinal P-glycoprotein in the interaction of digoxin and rifampin. J Clin Invest 1999;104:147–153.)
An additional consideration is that distribution of transporters along the gastrointestinal tract varies. Certainly, this is true for P-glycoprotein, with activity increasing down along the intestinal tract, being highest in the large intestine. This higher activity may, in part, explain why such drugs as ranitidine have such a much lower permeability in the colon than in the small intestine (see Fig. 7-2).
CAUSES OF LOSS IN ORAL BIOAVAILABILITY
The oral bioavailability (F) of drugs is commonly less than one, even when administered in solution. There are many reasons for the reduced systemic absorption, in addition to low intestinal permeability. Recall from Chapter 2, Fundamental Concepts and Terminology, that a drug must pass sequentially from the gastrointestinal lumen, through the gut wall, and through the liver, before entering the general circulation (Fig. 7-5). This sequence is an anatomic requirement because blood perfusing virtually all gastrointestinal tissues drains into the liver via the hepatic portal vein. Drug may also be lost by decomposition in the lumen; the fraction entering the intestinal tissues, FF, is then the fraction neither lost in the feces nor decomposed in the lumen. Of this permeating drug, only a fraction may escape destruction within the walls of the gastrointestinal tract, FG, thereby reducing the fraction of dose reaching the portal vein further to FF · FG. If drug is also eliminated in the liver, an additional fraction, FH, of that reaching the liver escapes extraction there, another site of first-pass loss. Accordingly, the measured overall oral systemic bioavailability, F, is then
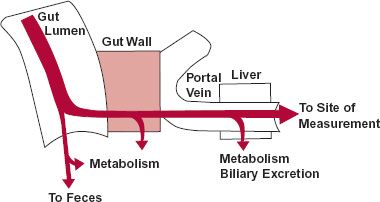
FIGURE 7-5. A drug, given as a solid or a solution, encounters several barriers and sites of loss in its sequential movement from the gastrointestinal tract to the systemic circulation. Dissolution, a prerequisite to movement across the gut wall, is the first step. Incomplete dissolution, slow penetration of the gastrointestinal membranes, or decomposition in the gut lumen are causes of poor bioavailability. Removal of drug as it first passes through gut wall and the liver further reduces the systemic bioavailability.
For example, if 50% of the drug is lost at each step, the bioavailability of the drug, measured systematically, would be 0.5 × 0.5 × 0.5 = 0.125, or 12.5%. Note that the drug can be rendered totally unavailable systemically at any one of these steps.
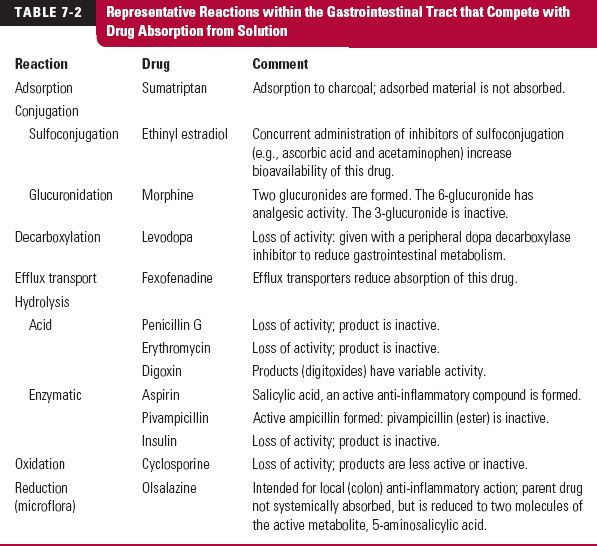
Competing Intestinal Reactions. Any reaction that competes with intestinal uptake reduces the oral bioavailability of a drug. Table 7-2 lists such reactions. They can be either enzymatic or nonenzymatic in nature. Acid hydrolysis in the stomach is a common nonenzymatic one. Enzymatic reactions include those caused by digestive enzymes (from bile and pancreatic fluids), metabolic enzymes within the intestinal epithelium, and microfloral enzymes, predominately resident in the large bowel. Complexation reactions with other drugs also occur; the result may be low drug bioavailability. For example, coadministration of activated charcoal or cholestyramine reduces the absorption of a number of drugs, which include the active metabolite of leflunomide, an agent used to treat rheumatoid arthritis; cephalexin, an antibiotic; and piroxicam, an analgesic agent. When both an adsorbent and an adsorbable drug are concurrently used, their administration must be appropriately timed to avoid their concurrent presence in any region of the gastrointestinal tract, particularly the small intestine. Otherwise, the bioavailability of the drug may be greatly reduced.
The complexities that occur in vivo make quantitative prediction of the contribution of a competing reaction to decreased bioavailability difficult. Sometimes, the problem of incomplete absorption can be circumvented by physically protecting the drug from destruction in the stomach or by synthesizing a more stable derivative, which is converted to the active molecule within the gastrointestinal tract or within the body. Similarly, to enhance absorption, more permeable derivatives are made, which are rapidly converted to the active molecule, often during passage through the intestinal wall. For example, absorption of the polar antibiotic ampicillin is incomplete. The systemic delivery of this acidic drug is improved substantially by administering it as a more lipophilic and permeable inactive ester prodrug, pivampicillin, which quickly releases ampicillin. Another example is that of valganciclovir (Valcyte), an antiviral agent. The hydrolysis of this compound by esterases within the gut wall and liver is so rapid that only its active metabolite, ganciclovir, is detected in the systemic circulation. Valganciclovir is therefore, by design, a prodrug as well.
First-Pass Loss. Metabolism during passage across the intestinal wall and through the liver reduces the amount reaching the general circulation. The drug is then said to undergo first-pass metabolic loss. A few examples to illustrate first-pass loss follow.
Aspirin (acetylsalicylic acid) is one of the first synthetic prodrugs. It was marketed at the end of the 19th century to help overcome the unpleasant taste and gastrointestinal irritation associated with the anti-inflammatory drug, salicylic acid. Aspirin was originally thought to be inactive, being designed to be rapidly hydrolyzed within the body to salicylic acid. Only subsequently was aspirin itself shown to have some pharmacologic effects of its own, namely an anticlotting effect. Yet the original design worked; upon ingestion, aspirin, a labile ester, is rapidly hydrolyzed, particularly by esterases in the gut wall and liver. Indeed, intestinal and hepatic hydrolysis is so rapid that a sizeable fraction of intestinally absorbed aspirin is converted to salicylic acid in a single passage through these organs, resulting in a substantial loss of oral bioavailability.
Another example of a drug showing first-pass loss is orlistat (Xenical). Apart from having a first-pass loss, one in the gastrointestinal wall (orlistat) and the other primarily in the liver (aspirin), orlistat and aspirin have little in common. They have different chemical structures and possess different pharmacologic activities. Aspirin (MW = 190 g/mol) is a simple acetyl ester of salicylic acid, whereas orlistat is a larger (MW = 496 g/mol), more complex molecule. Aspirin is an anti-inflammatory agent through its active metabolite, salicylic acid, whereas orlistat acts locally as a lipase inhibitor within the gastrointestinal tract to slow fat absorption and help control obesity. The almost complete first-pass metabolism of the small fraction of orlistat that permeates the intestine has, therefore, little impact on its efficacy.
When metabolites formed during the first pass through the intestinal wall and liver are inactive or less potent than the parent drug, the oral dose may need to be larger than the equivalent intravenous or intramuscular dose if the same therapeutic effect is to be achieved. Any drug with a high hepatic extraction ratio (see Table 5-2) has a low and often highly variable oral bioavailability. Being physiologically determined, no amount of pharmaceutical manipulation can improve on this value for an oral dosage formulation. Sometimes, the problem is so severe that either the drug must be given by a parenteral route, or it must be discarded in favor of another drug candidate. Flumazenil, a benzodiazepine receptor antagonist, and naloxone, an opioid antagonist, are examples. These drugs are so highly extracted by the liver that they must be given parenterally to be effective.
Extensive hepatic extraction can be advantageously used in therapy. Talwin (Hospira), a combination product of pentazocine, a potent narcotic analgesic with abuse potential, and naloxone, an analgesic antagonist, is effective as an analgesic when administered orally because naloxone, but not pentazocine, is very extensively metabolized during the first pass through the liver. However, when administered parenterally, the mixture is inactive because of the antagonistic effect of naloxone. The advantage of the combination in the oral product is to prevent its parenteral injection by drug abusers. Another combination product, Suboxone, containing naloxone and the narcotic analgesic buprenorphine, is given sublingually. Naloxone is also found not to be absorbed systemically when given by this route, but presumably a quicker response to buprenorphine is obtained by this dosage form than when given orally. Furthermore, the bioavailability of buprenorphine is greater after sublingual than after oral administration, probably because the vasculature in the mouth drains directly into the superior vena cava, thereby bypassing the liver.
Another example is budesonide, a synthetic corticosteroid used in treating Crohn’s disease of the ileum and ascending colon. The drug is given in a modified release dosage form which releases drug in the region where the disease is common. Drug easily permeates the intestinal wall but, owing to extensive first-pass metabolism in gut wall and liver, systemic availability is low, thereby reducing the adverse systemic effects of the corticosteroid. Coadministration of drugs that inhibit metabolism, however, reduces the first-pass loss and increases systemic exposure and therefore adverse events. Coadministration of ketoconazole, a potent inhibitor of metabolism at conventional doses, for example, has been reported to increase by eightfold the AUC of budesonide.
SEPARATING GUT WALL FROM HEPATIC FIRST-PASS LOSS
It is important to distinguish between gut wall and hepatic first-pass loss, as some effects, such as drug interactions to be discussed later in this chapter (see the “grapefruit juice–simvastatin interaction”), occur predominantly in the gut wall, others in the liver, and still others in a combination of both.
Consider an orally administered drug all of which enters the intestinal wall, that is FF = 1, so that F = FG · FH· An estimate of FG can be made if F and FH are known. F is determined experimentally, whereas FH may be calculated from the hepatic extraction ratio, EH, since

It is the maximum value of oral bioavailability if the only source of loss is hepatic extraction. Recall, from Chapter 5, Elimination, hepatic extraction ratio can be estimated if the hepatic (blood) clearance and hepatic blood flow are known or can be approximated.
To illustrate this, consider the following data obtained after oral and i.v. doses, each 5 mg, of a drug with a plasma-to-blood concentration ratio (C/Cb
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


