CHAPTER 16 Abnormalities in leukocyte morphology and number
Neutrophils
In the peripheral blood (PB) of healthy white adults the mean neutrophil count is 4.4 (4.3–4.6) × 109/l,1,2 while in healthy black adults it is 3.6 (3.3–3.9) × 109/l.3
Neutropenia
Neutropenia is defined as the absolute neutrophil count (ANC) in PB lower than 1.5 × 109/l. Referring to white population, a useful classification in predicting risk infection indicates neutropenia as mild, moderate or severe according to the ANC value of 1.0–1.5 × 109/l, 0.5–1.5 × 109/l or less than 0.5 × 109/l respectively. Neutropenia may arise from various disorders of bone marrow (BM) function resulting in a decreased rate of release of neutrophils from the BM into the circulation. It is a common manifestation in several BM failures, such as aplasia, leukemia or myelodysplasia (see Chapters 13, 18, 20 for details). The conditions that may be associated with a neutropenia are listed in Box 16.1. Clinically neutropenia is considered as acute when it occurs over hours to a few days, usually developing from rapid neutrophil use/destruction or from impaired production. Neutropenia is considered chronic, when it lasts months to years, usually arising from reduced production or excessive splenic sequestration. Neutropenia can be caused by an intrinsic defect in BM myeloid cells, the congenital and idiopathic neutropenias, or by factors extrinsic to BM, the acquired neutropenias.
Box 16.1 Main causes of neutropenia
Congenital and idiopathic neutropenias
Severe congenital neutropenia (Kostmann’s disease). Kostmann disease4 is a rare autosomal recessive disorder of neutrophil number. The ANC is characteristically less than 0.2 × 109/l. Severe persistent neutropenia results in an increased susceptibility to bacterial infections. Granulocyte colony-stimulating factor (G-CSF) receptors are expressed on myeloid cells in slightly increased numbers, while the binding affinity for G-CSF to its receptor is normal. The neutrophil elastase gene mutations (ELA2) have been found in a subgroup of patients with Kostmann disease, although these mutations are also found in some healthy family members.5 Autosomal recessive inheritance6 of homozygous hematopoietic cell-specific protein-1 (HS1)-associated protein X-1 (HAX-1) mutations appear to lead to the increased apoptosis of myeloid precursors observed in patients with severe congenital neutropenia.7 HAX-1 functions include signal transduction, cytoskeletal control and regulation of apoptosis and it is reported to play a role in suppression of apoptosis in lymphocytes and neurons, resulting in prolonged survival of these cells.8 While the G-CSF receptor gene mutations have not been detected at birth, patients with Kostmann disease who develop leukemia have been found to have acquired these mutations.9 Point mutations in the gene for the G-CSF receptor CSF3R have been implicated in the progression of severe congenital neutropenia to leukemia. These mutations lead to the production of truncated CSF3R receptors determining hyper-responsive forms (G-CSFR-hyper) with increased proliferation and highly decreased maturation of myeloid cells.10,11
Congenital aleukia (reticular dysgenesis). Reticular dysgenesis12 is a rare inherited disorder characterized by the failure of hematopoietic stem cells committed to myeloid and lymphoid development. Red blood cell count (RBC) is normal, while platelet count may be decreased. There are no lymphocytes in the thymus, which is hypoplastic, and there is complete absence of peripheral lymphoid tissues. Allogenic BM transplant (BMT) remains the sole therapeutic option.13
Benign chronic neutropenias
Familial benign chronic neutropenia. This disorder,14,15 inherited as an autosomal dominant trait, is characterized by chronic moderate to severe neutropenia, monocytosis, lymphocytosis and occasionally moderate eosinophilia. Patients with mild or moderate neutropenia are asymptomatic. The BM is normocellular and usually shows few neutrophil precursors beyond the myelocyte stage. Patients have a reduced reserve myeloid pool in the BM, which makes G-CSF therapy not appropriate.16
Non-familial benign chronic neutropenia. Cases of chronic benign neutropenia without any identified familial pattern are grouped in this category. In several patients, the presence of autoantibodies suggests the immune aetiology.17 Even though neutropenia is usually severe in patients with chronic benign neutropenia of infancy and childhood, the risk of infection is very low. BM is hypercellular and the granulopoietic differentiation is present until the stage of band form. A reduced neutrophil chemotaxis from BM to PB is a possible explanation. In situations causing acute stress such as infection or cortisol administration, the number of circulating neutrophils increases.18 G-CSF therapy in the forms with a benign course is not indicated.
Familial cyclic neutropenia. This is a rare disorder,19–21 in which neutropenia of 4–10 days duration occurs at intervals of 15–35 days (average 21 days). Most cases present in infancy or childhood with periodic bouts of fever, malaise, headache, sore throat, oral ulceration, skin infections or, occasionally, infections of the lungs and other organs. Neutropenia results from cyclic changes in neutrophil granulocytopoiesis. Cyclic neutropenia has been transferred by BM transplantation to a histocompatible sibling with acute lymphoblastic leukemia.22 In about 25% of patients, the condition is inherited as a dominant trait with point mutations involving the neutrophil elastase gene ELA-2 on chromosome 19p13.3. Sporadic, not familial, as well as acquired cases associated with large granular lymphocyte (LGL) expansion are reported in the literature.23,24
Chronic idiopathic neutropenia of adult. This is a cytokine-mediated syndrome characterized by varying degrees of neutropenia associated with a low number of lineage-specific CD34+ cells and increased production of inhibitors of hematopoiesis (including tumor growth factor (TGF)-β1 and tumor necrosis factor (TNF)-α) and lymphopenia due to selective loss of primed/memory T-cells and natural killer (NK) cells. Other symptoms, reported in about 50% of patients, include splenomegaly, osteopenia and/or osteoporosis. The presence of features of chronic antigenic stimulation and increased concentrations of a variety of macrophage-derived pro-inflammatory cytokines25 are suggestive of the existence of an unrecognized low-grade chronic inflammatory process, which may be involved in the pathogenesis of the disorder. Neutropenia in these patients is probably resulting from a combination of at least three factors:26 reduced production of BM neutrophils, enhanced neutrophil extravasation and increased sequestration and/or extravasation of neutrophils into the spleen.
Neutropenia in patients with congenital immune defects
Neutropenia can be associated with congenital immunologic defects and patients with combined defects are at very high risk of infection. Neutropenia is reported in about one fourth of patients with Bruton agammaglobulinemia (X-linked hypogammaglobulinemia),27 in about 50% of patients with dysgammaglobulinemia,28 and in several patients with hyper IgM syndrome or with isolated IgA syndrome.29 Treatment with intravenous infusion of immunoglobulin is required, possibly associated with G-CSF.
Neutropenia in patients with congenital chromosomal abnormalities
Shwachman–Diamond syndrome. Shwachman–Diamond30 syndrome is a rare autosomal recessive disorder characterized by pancreatic exocrine insufficiency, metaphyseal chondrodysplasia, mental retardation, thrombocytopenia and defective neutrophil motility. The mutations involve a single gene, the Shwachman-Bodian-Diamond-Syndrome (SBDS) gene on chromosome 7q11. Neutropenia is determined by an increased apoptosis of myeloid precursors caused by the SBDS. About 20% of patients evolve into acute leukemia.
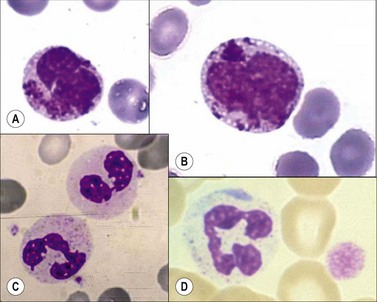
Chediak–Higashi syndrome. Chediak–Higashi31 syndrome is a rare autosomal recessive disorder characterized by recurrent infections, albinism, and by the presence of a reduced number of granules and/or formation of some abnormally large granules (by progressive fusion of normally formed granules) in most granule-containing cells. The giant granules present in neutrophils are peroxidase-positive and represent primary granules that have fused together. The abnormal granule fusion and function is determined by mutations of the CHS1 gene coding for the lysosomal trafficking regulator (LYST) protein on chromosome 1q42. Neutropenia is present in about 75% of patients. BMT provides a supportive cure for hematological signs but it does not prevent the neurologic deterioration.32 The affected cells include the neutrophils, eosinophils, lymphocytes, monocytes, melanocytes, Schwann cells of peripheral nerves, fibroblasts, vascular endothelial cells, renal tubular cells, and the parenchymal cells of the adrenal and pituitary glands. There is a deficiency of platelet dense granules, leading to easy bruising and bleeding. The large granules in leukocytes vary from a pale slate-gray to a dark reddish colour (Romanowsky stain) (Fig. 16.1A,B). There is a marked reduction in circulating NK cells.33 The leukocyte abnormalities are associated with recurrent infections. Some affected children, particularly those who live beyond early childhood, develop a terminal accelerated phase characterized by a lymphoma-like picture with lymphadenopathy, hepatosplenomegaly, neuropathy, widespread infiltration of tissues by non-clonal lymphatic and histiocytic cells, and pancytopenia. Death results from the complications of pancytopenia.
WHIM syndrome. Wart, hypogammaglobulinemia, infection and myelokathexis (WHIM) syndrome is a rare congenital autosomal dominant disorder determined by mutation in the gene coding for the neutrophil chemokine receptor CXCR.34 It is characterized by chronic non-cyclic neutropenia and increased susceptibility to bacterial and viral infections, especially from common serotype human papilloma virus, resulting in warts on the hands and feet starting in childhood. There is myeloid hyperplasia with degenerating granulocytes in the BM (myelokathexis).
Glycogen storage disease type 1b. It is a rare congenital autosomal recessive condition, determined by mutations on chromosome 11q23. These mutations cause glycogen accumulation in tissues due to deficiency of intracellular enzymes, which normally catalyze reactions that convert glycogen compounds to glucose. Severe neutropenia with consequent high risk of infections is present only in the type 1b and is determined by a disturbed myeloid maturation.35
Other rare conditions
Congenital dysgranulopoietic neutropenia. Congenital dysgranulopoietic neutropenia38,39is characterized by ineffective myelopoiesis and morphological abnormalities in the neutrophilic series. The term has been used to describe a disorder affecting six unrelated children presenting recurrent severe bacterial infections since birth, marked neutropenia, impaired neutrophil migration, normal numbers of BM colony-forming cells, normal or slightly increased colony-stimulating activity in the serum, and prominent morphologic and ultrastructural abnormalities in granulopoietic cells beginning from the neutrophil promyelocyte/myelocyte stage. The abnormalities of these cells included numerous autophagic vacuoles, abnormally electron-lucent primary granules, myelinization of primary granules, granule fusion, absence or marked decrease of secondary granules, and maturation of the cytoplasm ahead of the nucleus. Stem cell involvement in the pathogenesis of this disorder is postulated.40
Acquired neutropenias
Neonatal alloimmune neutropenia. In this syndrome, neutrophils and neutrophil precursors of the fetus are destroyed by the transplacental passage of maternal IgG alloantibodies formed against HLA or neutrophil-specific antigens (NA1 and NA2) present on fetal as well as on paternal neutrophils but not on maternal ones.41,42 The incidence is estimated at 1 in 500 live births. Most patients recover spontaneously usually within 2 months, depending on the life span of the maternal immunoglobulins. The neutropenia is occasionally severe and may cause life-threatening neonatal infections. Such patients respond well to intravenous immunoglobulins and G-CSF. The antibodies against neutrophil-specific antigens transferred to the fetus by the transplacental passage may be present either because the mother is affected by an autoimmune disease or as consequence of maternal sensibilization against the antigens of the fetal neutrophils of paternal origin. Despite a very low number of ANC, there is usually a spontaneous resolution within a few months. Supportive treatment with antibiotics is required in presence of infections, such as onphalitis or cellulitis.
Autoimmune neutropenia. Autoimmune neutropenia as a consequence of antineutrophil antibodies is a common finding associated with various collagen-vascular disorders, including rheumatoid arthritis (30% of patients with Felty’s syndrome), Systemic lupus erythematodes (SLE, 50% of patients), Sjögrens syndrome, scleroderma, polymyositis and polymyalgia rheumatica. It has also been described in association with autoimmune hemolytic anemia, immune thrombocytopenic purpura or both. In adults, autoimmune neutropenia is less likely to resolve spontaneously as compared to infants and children. In a subset of patients who have antibodies against the granulocyte precursors, the clinical course is more aggressive.43 Severe autoimmune neutropenias respond to treatment with metothrexate, steroids, G-CSF and granulocyte-macrophage colony stimulating factor (GM-CSF). Chronic autoimmune neutropenia44,45 is usually a relatively mild disease not associated with any other autoimmune disorders and characterized by recurrent infections, particularly of the oropharynx and skin. In some patients, the antineutrophil antibody has the specificity anti-NA2 or pan-Fcγ RIII (CD16, NA1/NA2). Some patients requiring treatment have responded to G-CSF. The LGL leukemia is often associated with neutropenia (see Chapter 28).
Infections associated with neutropenia
Infections are the most common cause of acquired neutropenia with different pathogenetic mechanisms (Box 16.1). In particular, some viruses (EBV, HAV, HBV, HCV and HIV) induce a severe and prolonged neutropenia by direct damage of the granulocytic precursors.46,47 Fungal, rickettsial and protozoal infections damage the BM endothelial cells causing vasculitis and severe cytopenias.48 Bacterial septicemia, in particular with Gram-negative agents, may induce neutropenia through an endotoxin-induced shift of neutrophils from the circulating to the marginated cell pools, a reduced survival time of circulating granulocytes (due either to destruction of cells within the circulation or to an accelerated rate of egress of cells from the blood), and a BM failure to adequately increase the rate of effective granulocytopoiesis. In certain circumstances, infection may be associated both with a failure adequately to increase effective granulocytopoiesis and with an exhaustion of the marrow granulocyte pool so that neutropenia is seen together with a left shift. This is particularly common in very severe bacterial infections and in bacterial infections in neonates and alcoholics (who have a reduced BM granulocyte pool). In several infections associated with spleen enlargement, such as typhoid fever, malaria and kala-azar, neutropenia results from splenic trapping and destruction of neutrophils in the spleen. In most of the latter cases, neutropenia does not require treatment, which should be focused on the underlying disease.
Neutropenia is the most common drug-induced blood dyscrasia.49,50 Some drugs (e.g. alkylating agents or antifolate drugs), certain chemicals (e.g. benzene) and irradiation induce neutropenia in all affected individuals in a dose-dependent manner. Other drugs cause neutropenia only occasionally and this phenomenon is usually at least partly based on a genetic polymorphism of drug metabolism. Drugs of the latter category may either cause neutropenia as part of an aplastic anemia (AA) or induce a selective neutropenia. Furthermore, several of the drugs causing AA in susceptible individuals initially cause a neutropenia that subsequently progresses to AA. The agranulocytosis induced by amidopyrin51 (and possibly also by other drugs) is often attributed to the destruction of circulating neutrophils and neutrophil precursors by drug-related immune mechanisms. There is also evidence that some drug metabolites may directly damage neutrophils and their precursors through the myeloperoxidase (MPO) system.52 Drugs that may occasionally cause selective neutropenia are listed in Box 16.2. The relatively high-risk drugs include amidopyrine and the antithyroid drugs. Some of the listed drugs usually induce a mild to moderate neutropenia that is asymptomatic and does not progress despite continuation of the drug. Other drugs usually cause a complete or almost complete absence of neutrophils (agranulocytosis).
Box 16.2 Main drugs which may cause neutropenia
Idiosyncratic drug-induced agranulocytosis (incidence 2.4–15.4 cases per million) results in a life-threatening clinical syndrome53 characterized by fever, sweating, vomiting, sore throat, dysphagia due to necrotic ulceration of the mouth and pharynx, extreme prostration and, frequently, death from overwhelming infection. At autopsy, necrotic ulcers are found in the mucous membranes of the entire alimentary tract as well as in the vagina. The prognosis is considerably improved if the offending drug is stopped and infection is adequately controlled by antibiotic therapy. To date, drug-induced agranulocytosis remains a serious adverse event due to the high frequency of sepsis with severe deep infections (such as pneumonia), septicemia, and septic shock in about two-thirds of all patients. Old age (>65 years), septicemia or shock, metabolic disorders such as renal failure, and a neutrophil count below 0.1 × 109/l are poor prognostic factors. Drugs or their metabolites might induce neutropenia by one or both of the following mechanisms: direct toxic effect on myeloid precursors or marrow environmental, dose-dependent inhibition of granulopoiesis, and immune-mediated destruction of granulocytes or their precursors. The classic example of a drug that causes agranulocytosis by a toxic effect on the BM is chlorpromazine, which causes a transient and moderate neutropenia in one-third of treated individuals. It causes agranulocytosis in about 1 in 1200 individuals, usually after a cumulative dose of 10–20 g over 20–30 days. Chlorpromazine appears to cause agranulocytosis by inhibiting DNA synthesis and cell proliferation in the granulocyte precursors of susceptible individuals. At the time of the agranulocytosis, the BM shows few or no neutrophil granulocytopoietic cells. The susceptibility of occasional individuals to develop agranulocytosis following therapy with chlorpromazine or other sulphur-containing drugs such as carbimazole or metiamide may depend on their genetically determined ability to oxidize the drug to highly-reactive myelotoxic metabolites (sulphoxides) more rapidly than most individuals.54 It is likely that many other drugs that cause neutropenia do so by directly or indirectly impairing biochemical processes within granulocytopoietic cells or by causing some form of immune destruction of the cells. The best understood example of a drug that causes agranulocytosis by destroying circulating neutrophils is amidopyrine. Individuals with amidopyrine-induced agranulocytosis have a history of previous exposure to the drug and may, after recovery from the initial agranulocytosis, show a recurrence of agranulocytosis within 12 h of a test dose. The serum of such individuals contains an antibody that causes agglutination of neutrophils followed by an acute neutropenia in the presence of the drug. It has been suggested that the antibody may be directed against complexes between drug metabolites and cellular components. The BM shows increased granulocytopoietic activity and a depletion of the more mature precursors. The dynamics of drug-induced neutropenia vary depending on the underlying mechanism: it has rapid onset in the immune-mediated mechanism, especially if the patient was previously exposed, while it is delayed when drugs exert direct marrow toxicity. Recombinant human G-CSF is indicated in patients who do not improve their ANC levels after the drug discontinuation.
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


