Chromosome abnormalities are described by a standard set of abbreviations and nomenclature that indicate the nature of the abnormality and (in the case of analyses performed by FISH or microarrays) the technology used. Some of the more common abbreviations and examples of abnormal karyotypes and abnormalities are listed in Table 5-1.
TABLE 5-1
Some Abbreviations Used for Description of Chromosomes and Their Abnormalities, with Representative Examples
Gene Dosage, Balance and Imbalance
For chromosome and genomic disorders, it is the quantitative aspects of gene expression that underlie disease, in contrast to single-gene disorders, in which pathogenesis often reflects qualitative aspects of a gene’s function. The clinical consequences of any particular chromosome abnormality will depend on the resulting imbalance of parts of the genome, the specific genes contained in or affected by the abnormality, and the likelihood of its transmission to the next generation.
The central concept for thinking about chromosome and genomic disorders is that of gene dosage and its balance or imbalance. As we shall see in later chapters, this same concept applies generally to considering some single-gene disorders and their underlying mutational basis (see Chapters 7, 11, and 12); however, it takes on uniform importance for chromosome abnormalities, where we are generally more concerned with the dosage of genes within the relevant chromosomal region than with the actual normal or abnormal sequence of those genes. Here, the sequence of the genes is typically quite unremarkable and would not lead to any clinical condition except for the fact that their dosage is incorrect.
Most genes in the human genome are present in two doses and are expressed from both copies. Some genes, however, are expressed from only a single copy (e.g., imprinted genes and X-linked genes subject to X inactivation; see Chapter 3). Extensive analysis of clinical cases has demonstrated that the relative dosage of these genes is critical for normal development. One or three doses instead of two is generally not conducive to normal function for a gene or set of genes that are typically expressed from two copies. Similarly, abnormalities of genomic imprinting or X inactivation that cause the anomalous expression of two copies of a gene or set of genes instead of one invariably lead to clinical disorders.
Predicting clinical outcomes for chromosomal and genomic disorders can be an enormous challenge for genetic counseling, particularly in the prenatal setting. Many such diagnostic dilemmas will be presented throughout this section and in Chapters 6 and 17, but there are a number of general principles that should be kept in mind as we explore specific types of chromosome abnormality in the sections that follow (see Box).
Unbalanced Karyotypes and Genomes in Liveborns
General Guidelines for Counseling
• Monosomies are more deleterious than trisomies. Complete monosomies are generally not viable, except for monosomy for the X chromosome. Complete trisomies are viable for chromosomes 13, 18, 21, X, and Y.
• The phenotype in partial aneuploidy depends on a number of factors, including the size of the unbalanced segment, which regions of the genome are affected and which genes are involved, and whether the imbalance is monosomic or trisomic.
• Risk in cases of inversions depends on the location of the inversion with respect to the centromere and on the size of the inverted segment. For inversions that do not involve the centromere (paracentric inversions), there is a very low risk for an abnormal phenotype in the next generation. But, for inversions that do involve the centromere (pericentric inversions), the risk for birth defects in offspring may be significant and increases with the size of the inverted segment.
• For a mosaic karyotype involving any chromosome abnormality, all bets are off! Counseling is particularly challenging because the degree of mosaicism in relevant tissues or relevant stages of development is generally unknown. Thus there is uncertainty about the severity of the phenotype.
Abnormalities of Chromosome Number
A chromosome complement with any chromosome number other than 46 is said to be heteroploid. An exact multiple of the haploid chromosome number (n) is called euploid, and any other chromosome number is aneuploid.
Triploidy and Tetraploidy
In addition to the diploid (2n) number characteristic of normal somatic cells, two other euploid chromosome complements, triploid (3n) and tetraploid (4n), are occasionally observed in clinical material. Both triploidy and tetraploidy have been seen in fetuses. Triploidy is observed in 1% to 3% of recognized conceptions; triploid infants can be liveborn, although they do not survive long. Among the few that survive at least to the end of the first trimester of pregnancy, most result from fertilization of an egg by two sperm (dispermy). Other cases result from failure of one of the meiotic divisions in either sex, resulting in a diploid egg or sperm. The phenotypic manifestation of a triploid karyotype depends on the source of the extra chromosome set; triploids with an extra set of maternal chromosomes are typically aborted spontaneously early in pregnancy, whereas those with an extra set of paternal chromosomes typically have an abnormal degenerative placenta (resulting in a so-called partial hydatidiform mole), with a small fetus. Tetraploids are always 92,XXXX or 92,XXYY and likely result from failure of completion of an early cleavage division of the zygote.
Aneuploidy
Aneuploidy is the most common and clinically significant type of human chromosome disorder, occurring in at least 5% of all clinically recognized pregnancies. Most aneuploid patients have either trisomy (three instead of the normal pair of a particular chromosome) or, less often, monosomy (only one representative of a particular chromosome). Either trisomy or monosomy can have severe phenotypic consequences.
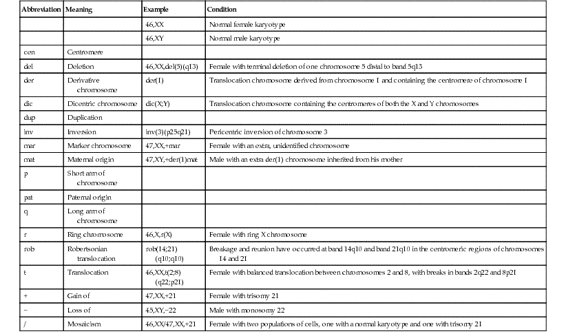
Trisomy can exist for any part of the genome, but trisomy for a whole chromosome is only occasionally compatible with life. By far the most common type of trisomy in liveborn infants is trisomy 21, the chromosome constitution seen in 95% of patients with Down syndrome (karyotype 47,XX,+21 or 47,XY,+21) (Fig. 5-9). Other trisomies observed in liveborns include trisomy 18 and trisomy 13. It is notable that these autosomes (13, 18, and 21) are the three with the lowest number of genes located on them (see Fig. 2-7); presumably, trisomy for autosomes with a greater number of genes is lethal in most instances. Monosomy for an entire chromosome is almost always lethal; an important exception is monosomy for the X chromosome, as seen in Turner syndrome (Case 47). These conditions are considered in greater detail in Chapter 6.
Although the causes of aneuploidy are not fully understood, the most common chromosomal mechanism is meiotic nondisjunction. This refers to the failure of a pair of chromosomes to disjoin properly during one of the two meiotic divisions, usually during meiosis I. The genomic consequences of nondisjunction during meiosis I and meiosis II are different (Fig. 5-10). If the error occurs during meiosis I, the gamete with 24 chromosomes contains both the paternal and the maternal members of the pair. If it occurs during meiosis II, the gamete with the extra chromosome contains both copies of either the paternal or the maternal chromosome. (Strictly speaking, these statements refer only to the paternal or maternal centromere, because recombination between homologous chromosomes has usually taken place in the preceding meiosis I, resulting in some genetic differences between the chromatids and thus between the corresponding daughter chromosomes; see Chapter 2.)
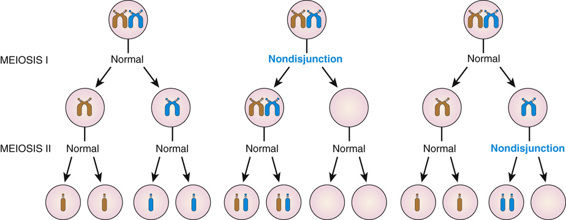
Proper disjunction of a pair of homologous chromosomes in meiosis I appears relatively straightforward (see Fig. 5-10). In reality, however, it involves a feat of complex engineering that requires precise temporal and spatial control over alignment of the two homologues, their tight connections to each other (synapsis), their interactions with the meiotic spindle, and, finally, their release and subsequent movement to opposite poles and to different daughter cells. The propensity of a chromosome pair to nondisjoin has been strongly associated with aberrations in the frequency or placement, or both, of recombination events in meiosis I, which are critical for maintaining proper synapsis. A chromosome pair with too few (or even no) recombinations, or with recombination too close to the centromere or telomere, may be more susceptible to nondisjunction than a chromosome pair with a more typical number and distribution of recombination events.
In some cases, aneuploidy can also result from premature separation of sister chromatids in meiosis I instead of meiosis II. If this happens, the separated chromatids may by chance segregate to the oocyte or to the polar body, leading to an unbalanced gamete.
Nondisjunction can also occur in a mitotic division after formation of the zygote. If this happens at an early cleavage division, clinically significant mosaicism may result (see later section). In some malignant cell lines and some cell cultures, mitotic nondisjunction can lead to highly abnormal karyotypes.
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


