Fig. 3.1
“Core” reactions that produce or remove reactive oxygen species
There remains debate among serious chemists (which this author makes no claim to be) as to which of the reactions in Fig. 3.1 are truly pathogenic and exactly what each of the “core” antioxidants evolved to accomplish (Linnane et al. 2007; Bartosz 2009; Gutteridge and Halliwell 2010; Sohal and Orr 2012). The fact that many therapeutics targeting ROS or RNS generally confer hearing protection (see below) certainly suggests that some reactions in Fig. 3.1 are best minimized. However, the mixed success record of such therapeutics indicates that Fig. 3.1 is missing too many elements, or too much spatial variation, to serve as a template for therapies. One entire class of peroxide-removing enzymes, the peroxiredoxins, may play a larger role than is generally credited (Rhee et al. 2005). As we noted, NO production is hugely dependent on the distribution of NOS isoforms, which vary by cochlear cell type. This may help explain an extremely confusing literature regarding the helpful-versus-harmful status of NO.
3.2.1 The “Purpose” of ROS and RNS Generation and Requirement for Balance
Without redox reactions, there would be no interesting or vital organic chemistry, so it is not as if life could be founded on some safer—less mischievous—set of reactions. Cells must be able to break down complex organic molecules and to kill invading pathogens. Both sets of reactions are oxidative. Eukaryotic cells produce superoxide as part of the mitochondrial respiratory chain. Environmental stressors, such as heat, osmotic stress, or pH imbalance, can enhance net superoxide production by mitochondria. Superoxide generation by phagocytes also occurs through the actions of a class of enzymes termed NADPH oxidases (NOX), particularly NOX2 (Bylund et al. 2010). However, nearly all cells produce one or more NOX isoforms, partly because most cell types retain some intrinsic ability to kill pathogens. NOX3 is especially highly expressed in the cochlea, where it may also participate in the formation of otoconia (Banfi et al. 2004). For reasons that are unclear, it is also highly expressed in hair cells and spiral ganglion cells, albeit minimally in the cochlear lateral wall. Under conditions of stress (e.g., noise and ototoxic exposure), NOX3 has been suggested to be the single largest producer of superoxide.
In addition to NADPH oxidases, cells produce a host of functionally similar enzymes residing in the cell membrane whose primary function may be generation of superoxide and peroxide to serve as messengers. Some have argued that most peroxide production is adaptive (Rhee 2006; Linnane et al. 2007; Sohal and Orr 2012). According to one line of logic (Bartosz 2009), any moiety that is created in an enzyme-catalyzed reaction serves a purpose. When the product is a potential oxidant, it will possess a narrow non-pathological concentration range. Moreover, the enzymes that make the product must be balanced by other enzymes that remove it from the same compartment (Rhee 2006). Many transcription factors and other mediators that drive cell processes in non-stress contexts (e.g., division, migration, secretion, and contraction) are redox-sensitive, including AMP-activated protein kinase (AMPK), platelet-derived growth factor (PDGF), p38 MAP kinase, c-Jun N-terminal kinase (JNK), and nuclear factor kappaB (NFκB) (Veal et al. 2007; Bartosz 2009). In such cases, transcriptional activation relies on reversible modifications to transcription factors, cofactors, and binding sites. Under conditions of overt stress (heat, osmotic stress, pH change, shear stress, hypoxia, hyperoxia, infection), oxidant concentrations may approach the pathological range and engage mediators of antioxidant defenses and repair-versus-die “decisions.” These overlap with the previous set and include hypoxia-inducible factor 1α (Hif1α), Nrf-2, Atf1, JNK, and NFκB. Injury to excitable cells (as in the brain and cochlea) can be modulated by altering ion conductances, particular to K+ and Ca++. Thus it is no surprise that voltage-activated and gap junctional conductances are readily altered by oxidation, often (not in all cases) in such a way as to dampen activation (Bartosz 2009).
In the inner ear, peroxide production by SOD and removal by catalase and GPx appear to operate in critical balance. Targeted inactivation of GPx1 and SOD1 in mice was found to accelerate age-associated hearing loss and exacerbate the effects of noise (McFadden et al. 1999a, b, 2001; Ohlemiller et al. 1999, 2000), but overexpression of SOD1 was found to be harmful to the cochlea (Coling et al. 2003). How toxic is peroxide to the inner ear? Experiments attempting to demonstrate peroxide toxicity in the cochlea (Clerici et al. 1995; Clerici and Yang 1996) and other tissues (Linnane et al. 2007; Sohal and Orr 2012) have used nonphysiologically relevant high levels, so that these may not be very informative. Purely pathologic functions of peroxide may result from its conversion to hydroxyl radical through reactions with iron via Fenton/Haber–Weiss chemistry (Fig. 3.1). Hydroxyl radical seems to have no adaptive role in cell physiology. However, it is so reactive—reacting with the first molecule it encounters—that some have expressed doubts that it is truly biologically relevant (Linnane et al. 2007).
Knockout mouse models generally support the notion that impairment of the “core” antioxidants of Fig. 3.1 can produce individuals that are susceptible to environmental stress (Yoshida et al. 1997; Lei 2001; Ho et al. 2004). This principle appears to extend to acquired hearing loss in humans. Certain single nucleotide polymorphism (SNP) variants of genes encoding a number of redox-related enzymes, including catalase, SOD2, paraoxonase-2 (PON2), and glutathione-S-transferase M1 (GSTM1), have been implicated in decreased resistance to permanent threshold shifts (PTS), particularly in the context of habitual noise exposure (Sliwinska-Kowalska and Pawelczyk 2013) (see also Chaps. 8 and 14). GSTM1 genotypes associated with increased hearing loss in humans included homozygous nulls—that is, individuals who do not produce this enzyme at all. Such results are relevant to how we conceptualize and search for “pro-PTS” alleles. It might be argued that testing candidate genes in homozygous null (knockout) animals represents an unrealistic and biased way to study such genes, since alleles contributing to PTS in humans will typically not be nulls nor will they be inherited in a homozygous state. However, SNP variants are limited to just four allele types (since there are only four possible bases), of which subsets will be enriched in certain populations, and thus are likely to be inherited in homozygous form. The GSTM1 results further argue that some naturally occurring variants may, in fact, be null alleles. Most of the current evidence for pro-PTS alleles in humans is tentative, as most studies used small samples and very few have been replicated. Part of the difficulty is that relevant human gene variants are likely to adhere to “common variant” notions, whereby alleles of many genes that are common across populations each contribute only a small amount of risk.
The set of reactions falling under the rubric of nitrosative stress begins with the generation of nitric oxide (Berg et al. 2004; Heinrich and Helling 2012). Through reaction with superoxide, NO promotes the production of peroxynitrite (Radi 2013). Nevertheless, adaptive functions have been proposed for both NO and peroxynitrite. NO is produced by distinct synthase isoforms whereby it supports neuronal synaptic function (through the activity of neuronal NOS, nNOS) and promotes increased numbers of mitochondria and vasodilation (through endothelial NOS, eNOS). Maladaptive generation of NO is driven most often by stress-related activation of inducible NOS (iNOS), which is minimally detectable in the cochlea under normal conditions (Shi et al. 2003), but can produce much higher amounts of NO than the other isoforms. Notably, the primary activity of iNOS in the body occurs in macrophages in response to pathogens or exposure to lipopolysaccharide (LPS, a component of bacterial cell walls), so that iNOS-related nitrosative stress represents an aspect of inflammation. Within the cochlea, LPS, noise, and ototoxins can all upregulate iNOS (Heinrich and Helling 2012).
Peroxynitrite, the single major RNS in much of the literature, is moderately membrane permeant and may diffuse several cell diameters before reacting with proteins, lipids, and nucleic acids (Radi 2013). The reaction of NO with superoxide to form peroxynitrite is much faster than the reaction of superoxide with SOD. Hence, local production of peroxynitrite is highly sensitive to the joint concentration of superoxide and NO.
3.2.2 What Evolutionary Forces Shaped the Stress Responses of the Inner Ear?
It is tempting to think of evolution as a pure “optimizer” (through selective pressure) of how cells and organisms respond to their environment. It is questionable, however, how strongly selective pressures could have acted to optimize responses of cochlear cells to industrial age noise or ototoxins (for discussion, see Kirk and Smith 2003). It makes sense that mechanisms (e.g., cochlear efferents) evolved to impart gain control and to optimize signal detection in noise and that these may be co-opted to modulate noise injury. But apparent gaps in regulatory systems, and some clearly maladaptive responses of the cochlea to stress, conjure a vision of evolution more like a toddler with a jackhammer than a skilled mechanic. Innate shortcomings that now leave researchers trying to play the role of optimizer are likely the result of selection for one feature working at cross-purposes to other necessary features that may not be obvious. For example, hair cell self-repair seems limited, yet hair cells convinced not to die by anti-apoptotic treatments show surprising capacity for self-repair (Pirvola et al. 2000). Why is this not the natural response? A variety of preconditioning paradigms are highly effective in protecting hair cells from noise (e.g., Canlon 1997; Yoshida et al. 1999; Yoshida and Liberman 2000; Gagnon et al. 2007; Fernandez et al. 2010), yet there must be a downside to maintaining the preconditioned state or it would always be engaged. A frequent evolutionary driver for preconditioning was probably bacterially and virally derived endotoxin (Tsai et al. 2004). This would help explain why the response of the inner ear to virtually any stressor we invoke somewhat resembles a response to pathogens. In both noise- and ototoxin-exposed cochleas, hair cells, supporting cells, and cells of the lateral wall secrete a host of cytokines. The actions of these are neither unambiguously good nor bad. Chief among their actions is the apparent recruitment of inflammatory cells from the vessels of the spiral ligament (Hirose et al. 2005; Tornabene et al. 2006). Following noise, LPS, or ototoxins, multiple distinct types of macrophages (as determined by their surface receptors) invade the ligament and the lining of the scala tympani. By and large, they seem to migrate to sites of cell loss (spiral ligament and limbus) and may primarily simply be clearing debris. Yet they rarely invade the organ of Corti, even when there is extensive hair cell loss. Within the organ of Corti, their debris-clearing role may be supplanted by resident macrophages (Abrashkin et al. 2006). Even for macrophages that remain within the ligament, it seems to matter what types of cell surface receptors they express. Sautter et al. (Sautter et al. 2006) showed that mice lacking chemokine receptor 2 (CCR2, a particular macrophage surface receptor) may sustain more noise-induced PTS than wild-type controls. This suggests there exists some type of communication between these cells and the organ of Corti. Alternatively, if the macrophages act solely to preserve the spiral ligament, the results are consistent with dependence of the organ of Corti on the condition of the ligament.
3.3 Hair Cells as Generators and Targets of ROS/RNS
3.3.1 Noise and Hair Cells
It is actually not obvious why excessive noise should impose ROS/RNS stress on cochlear hair cells. At least for sub-traumatic exposures, all hair cells have to do is passively gate transducer currents through their apical and lateral membranes. Even the outer hair cell (OHC) motility “amplifier” utilizes the regenerative simplicity of a molecule (prestin) that transitions between two conformations, without adenosine triphosphate (ATP) hydrolysis (Dallos et al. 2006). The limiting ability is likely to be maintenance of appropriate Ca++ levels. Calcium enters the cell through the transducer channel where it modulates the adaptation state of the hair bundle (Ceriani and Mammano 2012) and is removed by ATPase pumps, also within the stereocilia. These pumps may be overwhelmed during noise exposure. In active hair cells, intracellular Ca++ levels increase from multiple sources, including release from Hensen’s bodies and influx through multiple membrane channels, including ATP-gated Ca++ currents in the hair cell lateral membrane. The increased Ca++ drives glutamate release from inner hair cells (IHCs) and modulates somatic contractions of OHCs. Yet the allowable range for intracellular Ca++ is very narrow. For reasons that hardly seem adaptive, excess Ca++ in hair cells is taken up by mitochondria, where it can trigger increased and unbalanced superoxide and NO production (Shi et al. 2003; Shi and Nuttall 2003). The most straightforward action of Ca++ may be to alter mitochondrial membrane potential through accumulation of positive charges (Arundine and Tymianski 2004). Böttger and Schacht (Böttger and Schacht 2013) further suggest that a particularly compelling point of intersection between Ca++ and mitochondrial function is the Ca++-modulated Krebs cycle enzyme α-ketoglutarate dehydrogenase. Compromised mitochondria may reach a threshold “permeability transition,” at which point they release cytochrome C, apoptosis-inducing factor (AIF), and endonuclease G and engage various caspases, tipping the balance toward apoptosis (Han et al. 2006). Aside from its effects on mitochondria, cytoplasmic Ca++ engages proteases and phospholipases, with subsequent release of prooxidant arachidonic acid. The role of Ca++ in hair cell injury is supported by the benefits of both L- and T-type Ca++ channel blockers against noise- and age-related hearing loss in mice and perhaps humans (Shen et al. 2007; Uemaetomari et al. 2009).
A substantial literature highlights the value of cochlear pharmacotherapies targeted against both ROS and RNS (for reviews, see Le Prell et al. 2007; Ohlemiller 2008; Le Prell and Bao 2012), clearly implicating these in hair cell injury and supporting the feasibility of this approach (see also Chaps. 9, 12, and 16). The role of NO in hair cell injury is perhaps a bit ironic, in that one major role of nNOS (the major hair cell isoform) is taken to be negative feedback on Ca++ entry (Shen et al. 2003). NO may readily exceed adaptive levels, partly because iNOS is also activated (Yamamoto et al. 2009). Besides giving rise to peroxynitrite, it overactivates PARP1 (poly-ADP-ribose polymerase-1), a DNA repair enzyme that depletes cellular ATP and NAD+ energy currency in the process (Schreiber et al. 2006).
How can the protective systems of cochlear cells become so easily unbalanced and maladaptive? The conditions we create in the laboratory, cochlear cellular stress responses simply did not evolve to resist. What we create in the lab—or perhaps in recreational and occupational settings—were never experienced by cochlear cells throughout evolution. Since we have no “head-to-head” comparison of the hardiness of human and animal hair cells, we should be prepared for fundamental differences in the investment different species may make in hair cell survival or repair. Indeed, a host of studies suggest intrinsic differences in vulnerability to noise. Susceptibility to noise-induced PTS varies across inbred strains of mice (e.g., Yoshida et al. 2000; Davis et al. 2001) and between rodent species. CBA/Ca mice and albino Sprague–Dawley rats appear more vulnerable than pigmented guinea pigs (Duan et al. 2008), which appear more vulnerable than humans (Liang 1992). Notably, this calculus takes into account only hearing thresholds: For a given amount of PTS, CBA/Ca mice actually lose fewer hair cells than do rats, guinea pigs, and perhaps humans (Ohlemiller 2012). The surviving cells are perhaps simply not repaired (Wang et al. 2002).
3.3.2 Ototoxicants and Hair Cells
The primary cochlear targets of aminoglycoside antibiotics are hair cells, particularly OHCs (Poirrier et al. 2010; Xie et al. 2011; Heinrich and Helling 2012). This does not appear to reflect greater uptake by hair cells, but rather is particular to the biology of hair cells versus non-sensory cells. Generation of ROS in hair cells following gentamicin has been demonstrated using fluorescent or other indicators (Clerici et al. 1996; Hirose et al. 1997; Jiang et al. 2005). Aminoglycosides can directly catalyze ROS formation, as well as activate iNOS, but also appear to cripple antioxidant protections. As in the case of noise, mitochondrial injury and NOX activity play a major role in ototoxicant-related oxidative stress, and generally similar cascades trigger apoptosis. By contrast with noise (Yamamoto et al. 2009), NFκB appears to play a more unambiguous role in endogenous protection. Iron chelators and a wide range of antioxidants have proven effective against aminoglycoside-induced hair cell loss (Le Prell et al. 2014).
Cellular targets and ototoxic mechanisms of the antineoplastic cisplatin overlap with those for aminoglycosides, except that cisplatin may more aggressively deplete cellular antioxidants and engage more NOX isoforms. It may also more readily promote hair cell Ca++ entry via transient receptor potential vanilloid-1 (TRPV1) channels (Mukherjea et al. 2008) (see also Chaps. 10 and 11).
3.3.3 Aging and Hair Cells
We are born with cochleas possessing an excess of strial and ligament pumping capacity and, to some degree, redundant afferent neurons. Regarding hair cells, if only IHCs and behavioral hearing thresholds are considered, we may be able to tolerate up to 80 % losses (Lobarinas et al. 2013). However, thresholds appear fairly intolerant to OHC losses (Hamernik et al. 1989). Mammalian cochlear hair cells die once and do not regenerate. Each day, they are confronted anew with the decision whether to die and must “decide” based on a balance of factors. While one factor just might be the host’s taste in music, the bulk of the decision likely rests on the cells’ overall mitochondrial status. That is, to what extent are “enough” mitochondria functional? Over the course of evolution, most mitochondrial genes have migrated to the nucleus. The remnant mitochondrial genome is small, fragile, poorly protected, and under-repaired (Böttger and Schacht 2013). Time imposes wear and tear from noise and ototoxicants, yet also from radiation and stray reactions, so that cumulative mitochondrial DNA damage is central to many notions of aging (Sohal and Orr 2012). What then follow are events similar to the sequelae of a noise exposure: a series of oxidative cascades and injury to lipids, proteins, and DNA. Human temporal bones reveal that presbycusic ears tend to be ears with more damage to mitochondrial DNA (Bai et al. 1997). Energetically disadvantaged cells do everything more poorly, including a poorer job of maintaining antioxidant defenses. The most significant lost capacity of old postmitotic cells may be defined as lost “operating margin”: reduced ability to respond to stress and a more precarious homeostatic balance. Extension of this operating margin is central to the touted advantages of caloric restriction and its mediators (Ohlemiller and Frisina 2008; Someya et al. 2010; Han and Someya 2013) (see Chaps. 13 and 16). Cochlear oxidative imbalance has been diagnosed in aging animals (Jiang et al. 2006), and antioxidant genes may be notably up- or downregulated with age in a manner consistent with chronic oxidative stress (Lautermann et al. 1997; Staecker et al. 2001; Tanaka et al. 2012). At present, evidence for the value of long-term pharmacotherapies against presbycusis is mixed (Poirrier et al. 2010; Heman-Ackah et al. 2010; Böttger and Schacht 2013).
3.4 Cochlear Afferent Synapses as Generators and Targets of ROS/RNS
3.4.1 Noise-Related Synaptopathy?
Until recently, cochlear noise injury was largely a hair cell story. Permanent threshold shifts were compared with hair cell loss, with general correspondence between the two. Transient swelling of the inner hair cell afferent synapses was a curious detail, perhaps with some role in temporary threshold shifts (TTS) (Pujol et al. 1991, 1993). Now, quite a stir has been raised over the discovery in mice that noise that induces a robust TTS can yield permanent synaptic injury that effectively de-afferents IHCs and promotes progressive neuronal loss (Kujawa and Liberman 2006, 2009). These effects can occur independently of hair cell loss. The functional effects do not manifest in threshold shifts, but rather in suprathreshold decreases in evoked potential amplitudes. If the basic findings apply to humans, expected perceptual correlates might include degraded speech reception in noisy backgrounds. However, the extent of this pathology, termed “synaptopathy,” varies by species, being less pronounced in guinea pigs and rats than in mice (Liu et al. 2012; Shi et al. 2013), and its applicability to humans remains speculative. One reason it may not readily translate is that the human cochlear nerve is anatomically distinct from the other species mentioned with regard to myelination and the presence of gap junctions between the somata (Tylstedt et al. 1997; Tylstedt and Rask-Andersen 2001; Glueckert et al. 2005). These features could increase sound-driven activity and promote the ultimate survival of human afferent neurons, even if they have been disconnected from their hair cell targets. Additional reasons for doubt derive from comparative neuronal counts in aged temporal bones of humans and mice (Makary et al. 2011; Sergeyenko et al. 2013). The similar trends by species—suggesting a linearly progressing loss of afferent neurons over the life-span in each—seem at odds with an expected skewing of human counts to lower values. The “absent” data points would represent the effects of undiagnosed noise exposure in the human sample. The verifiably unexposed mouse population would not display such an effect.
3.4.2 Synaptopathy and Aging
Even if not related to noise exposure, the notion of synaptopathy nevertheless fits with recognized features of aging temporal bones (Makary et al. 2011; Sergeyenko et al. 2013) and with aging trends in hearing. The single predominant complaint offered by the aged about their hearing is that environmental sounds (i.e., speech) are plenty loud, but the signal cannot be extracted from the noise (Frisina 2001, 2009) (see also Chap. 14). In the most severe cases, a diagnosis of neural presbycusis may be applied, but some degree of deficit may be nearly universal.
3.4.3 Mechanisms of Synaptopathy
What may drive the loss of afferent neurons whose target cells are still intact? The initial injury may involve excitotoxic effects of glutamate, which are oxidative and nitrosative in nature (Rego and Oliviera 2003; Gu et al. 2010; Kostandy 2012), and recapitulate the Ca++-based injury processes described above for hair cells. All glutamate receptor subtypes operating at the IHC afferent synapse (AMPA/kainate, NMDA) can contribute to this process. Ironically, the excitotoxic cascade likely involves NO generated by nNOS that is “intended” to protect against excess Ca++, yet which imparts net injury. Another surprising aspect of this story relates to NFκB. Following noise exposure, NFκB may contribute to broad injury of the organ of Corti and lateral wall through inflammation (Yamamoto et al. 2009) (for detailed discussion, see Chap. 19). At the IHC/afferent synapse, however, NFκB appears to serve as a brake on Ca++ entry. NFκB knockout mice show accelerated loss of afferent synapses fitting the pattern of synaptopathy (Lang et al. 2006). While synaptopathy may involve an initial stage that is excitotoxic, it may require additional processes that irreparably sever trophic signals that support dendritic connection and neuronal survival.
3.5 The Cochlear Lateral Wall as Generator and Target of ROS/RNS
3.5.1 Homeostasis of Endolymph and Perilymph Composition
The intracellular-like fluid that fills the scala media is termed endolymph, while the extracellular (and CSF-like) fluid that fills the scala tympani and vestibuli is termed perilymph. Neither endolymph nor perilymph appreciably “flow,” nor are they “pumped” (Patuzzi 2011a, b). Instead, water passively adjusts to ion gradients that are established locally, largely by the action of the stria vascularis. Cells of the organ of Corti that insert into the reticular lamina are bathed in endolymph at their apical surface and by perilymph on all other surfaces (Fig. 3.2). All nutrients must therefore reach these cells via one of these fluids, and all waste must be carried away by one of these. Endolymph, while serving as the critical source of K+, is generally not held to be the major source of nutrients nor the means by which metabolic waste is removed. Instead, perilymph must fill this need. Perilymph also serves as the milieu of the intercellular spaces of the spiral ligament and so is in free communication with the capillaries of the ligament. While tissues are typically composed such that most cells lie within 20 μm of a capillary, in the cochlea this distance may easily exceed 100 μm. This seeming problem, created perhaps by the need to separate mechanosensitive receptor cells from “noisy” capillaries, appears largely solved by a combination of factors. First is the evolutionary innovation of moving much of the work of pumping critical ions up their electrical and concentration gradients from hair cells to the lateral wall. This allows hair cells to operate at a lower metabolic rate than even non-sensory cells (Nakai and Hilding 1968). Another factor may be reliance on a network of gap junctions that connect nearly all cells lateral to the OHCs, through the inferior ligament, to the superior ligament, and finally to the cells of the stria (Wangemann 2006; Zhao et al. 2006; Nickel and Forge 2008). This web of connected cells is discontinuous only within the inferior ligament where ions and macromolecules released by outer sulcus/root cells (the epithelial junctional network) must again be actively taken up by type II fibrocytes (the connective tissue junctional network). Yet the web extends to the stria itself, ultimately linking type I fibrocytes of the ligament to strial basal and intermediate cells. This arrangement promotes K+ recycling to the stria yet also offers the possibility that both strial and ligament capillaries could support organ of Corti function. Indeed, work in connexin 30 (Cx30) mutant mice has revealed transfer of glucose from the stria to the ligament that is abolished in the mutants (Chang et al. 2008) (Fig. 3.5). Traffic though the gap junctional network must be two way, presumably with K+ and metabolic waste diffusing away from the organ of Corti and oxygen and glucose diffusing toward the organ. This traffic may be gated to some extent by ATP, inositol triphosphate (IP3), and Ca++ (Zhu and Zhao 2010). Both peroxide and NO reduce gap junctional conductance, so that injury from oxidative stress may be amplified through simultaneous effects on both the organ of Corti and lateral wall. It has long been clear that chronic dysfunction of lateral wall gap junctions promotes degeneration within the organ of Corti (Nickel and Forge 2008; Xu and Nicholson 2013). Mutations of connexin 26 (Cx26) account for roughly half of all cases of nonsyndromic hereditary deafness and are associated with such degeneration. The potential significance of transient disruption of gap junctions due to noise, ototoxins, or inflammation for permanent hearing loss is an emerging idea that may heighten interest in acute lateral wall injury (see below).
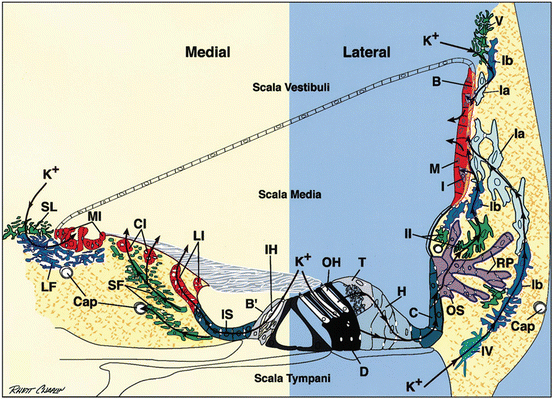
Fig. 3.2
Schematic radial view of the organ of Corti and adjacent lateral wall. Arrows indicate posited medial and lateral transcellular routes for K+ effluxed from inner and outer hair cells (IH, OH) during auditory transduction. B strial basal cell, B’ border cell, Cap capillary, C Claudius cell, CI central interdental cell, D Deiters cell, H Hensen cell, I strial intermediate cell, IS inner sulcus cell, M strial marginal cell, MI medial interdental cell, LF light fibrocyte, LI lateral interdental cell, OS outer sulcus cell, RP root process of outer sulcus cell, SF stellate fibrocyte, SL supralimbal fibrocyte, T tectal cell, Ia, Ib, II, IV, and V types of spiral ligament fibrocytes (From Spicer and Schulte 1998. Reprinted with permission Elsevier Publishing)
3.5.1.1 Limitations Posed by Vasculature
Distinctly different vascular loops serve Rosenthal’s canal and spiral ganglion cells versus the lateral wall and organ of Corti (Axelsson 1988; Axelsson and Ryan 2001). Capillaries of the stria and spiral ligament also likely serve the metabolic needs of the organ of Corti. The primary route is probably through the ligament to the lateral organ or through the scala tympani to the organ of Corti. The nutritive needs of the medial organ of Corti (particularly IHCs) may also be met by capillaries in the spiral limbus. The vessel of the basilar membrane is not maintained in adulthood in most non-primates, and its appearance even in primates is highly variable; thus it is unlikely to serve as the primary source of blood flow to the organ of Corti.
It is widely supposed that the sheer distance of the blood supply from hair cells presents a problem under demanding conditions such as noise exposure. Moreover, noise exposure appears to reduce cochlear blood flow via both inflammatory and oxidative mechanisms (Miller et al. 2003; Nakashima et al. 2003; Arpornchayanon et al. 2013). Noise-related vasoconstriction does not seem to occur within the stria. Rather it may occur in the superior ligament, in the type V fibrocyte region (Dai et al. 2011) (Figs. 3.2 and 3.3) where vessels that feed the stria and ligament emerge from the boney cochlear turn boundaries. This may explain why variation in blood flow (velocity) can be detected in the stria, yet also how vasoactive agents (e.g., TNFα, 8-iso-prostaglandin F2α) that are likely generated within the cochlea can nevertheless impact strial blood flow. Some capacity for local regulation may also exist within the capillaries of the ligament (Wu et al. 2011). The precariousness of the vascular supply to the organ of Corti may represent the single overarching weakness that exposes the organ to injury (see also Chap. 8).
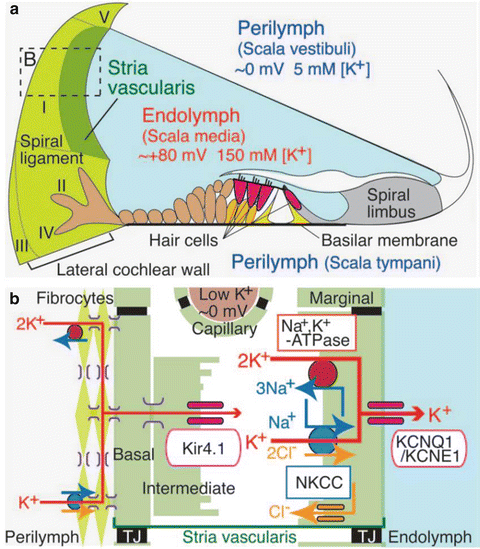
Fig. 3.3
Functional schematic structure of the cochlear duct with the lateral wall. (a) Appropriate ionic composition of the endolymph and the EP require an ion barrier lining scala media, the intrastrial space, and strial capillaries. A nearly continuous cellular network guides K+ from the organ of Corti through finger-like root cells into (primarily) type II and I fibrocytes. Type II fibrocytes must take up K+ from the extracellular space around root cells. Five major types of fibrocytes are indicated by roman numerals. (b) Enlargement of the boxed area in A depicts the cells and components needed to generate the EP. Type I fibrocytes, strial basal cells, and intermediate cells are joined by gap junctions composed mostly of connexins 26 and 30. K+ enters the intrastrial space through Kir4.1, then through marginal cells via Na+/K+-ATPase, the NKCC ion exchanger, and KCNQ1/KCNE1 channel complexes. TJ tight junction (Reprinted with permission from Nin et al. 2008)
3.5.2 Role of the Stria Vascularis in Hearing
After K+ has served its primary function of mediating hair cell sound responses, it is released into the fluid space around hair cells. Na+/K+ ATPases in both the type II fibrocytes of the spiral ligament and strial marginal cells create critical sinks for K+ recycling (Hibino and Kurachi 2006; Hibino et al. 2010) (Fig. 3.3). The type II fibrocytes create the first sink by lowering extracellular K+ levels at the root cell/type II interface. This yields a concentration gradient for K+ that facilitates its movement from the organ of Corti, through the epithelial gap junctional network, to the root cells. Once the type II fibrocytes have taken up K+, it is not actively pumped through the connective tissue gap junctional network, but rather moves down the concentration gradient created by the sink within the intrastrial space, created by strial marginal cells. The stria creates both the high, positive, endocochlear potential (EP) (+90–100 mV) and the high K+ levels of the scala media. Cochlear neuronal response thresholds and spontaneous activity, in turn, depend on the EP (Patuzzi 2011b). As might be expected, factors that reduce the EP, such as aging, and events such as aminoglycoside exposure, noise exposure, and loop diuretics, elevate physiological thresholds with a dependence of 0.5–1.0 dB/mV (Sewell 1984; Schmiedt 1993; Ohlemiller 2009) (Fig. 3.6). With the exception of aging and noise exposure in some strains and ages of mice (Ohlemiller et al. 2011), EP reduction generally appears temporary.
3.5.2.1 Requirements for an Endocochlear Potential
The essential ingredients of EP generation revolve around the availability of K+, the machinery to move K+, and the tight compartment boundaries to keep K+ corralled in the appropriate space.
Machinery for EP Generation
In the same way that an automotive fuel pump cannot work if the gas tank is empty, the machinery of EP generation must have K+ to work with. There must be an intact connexin 26, 30, and 31 gap junctional network for K+ to move from the organ of Corti, and type II fibrocytes must have functioning Na+/K+ ATPase to be able to take up K+ in the inferior spiral ligament. In Cx26 knockout mice, the EP is abnormal and hair cells degenerate, yet the stria retains a normal appearance.
The strial machinery for EP generation is shown in Fig. 3.3 (Hibino and Kurachi 2006; Wangemann 2006; Nin et al. 2008; Hibino et al. 2010). K+ is conveyed from spiral ligament into strial basal cells and intermediate cells by gap junctions. K+ then exits the intermediate cells into the intrastrial space via KCNJ10 (Kir4.1) channels, driven by the steep gradient for K+ that normally exists across this boundary. It is this step—large K+ flux across a high resistance—that produces the EP. This flux would not occur, however, if K+ were not readily taken up into marginal cells via Na+/K+ ATPase and the Na+/K+/Cl− exchanger. K+ is then released into the scala media via channels jointly formed from KCNQ1 and KCNE1. Note that as a consequence of the trilaminar construction of the stria, the generation of the EP and the establishment of high K+ levels are accomplished in separate layers and actions. The situation in the cochlear scala media contrasts with that in the endolymph of the utricle, where a monolayer of marginal cell-like dark cells produces a high K+ concentration but a very low EP.
Endolymphatic Boundaries
The boundaries of the scala media separate high-K+ endolymph from low-K+ perilymph and thus must be impermeable to uncontrolled ion flow. These boundaries are composed of all the cells that line the scala media, from the stria, to Reissner’s membrane, to the luminal surface of the spiral limbus, to the reticular lamina, and then finally to the outer sulcus cells and spiral prominence (Figs. 3.2 and 3.3). Also, since the intrastrial space must maintain very low K+ levels, tight junctions join marginal cells on the luminal side of the stria and basal cells on the abluminal side, so that this space is also ion-tight. Thus an additional set of boundaries surrounds the stria itself, setting off a fluid space that differs in composition from both endolymph and perilymph (Wangemann 2006). Finally, the endothelial cells that line strial capillaries are assumed to establish an ion-tight boundary that separates intrastrial fluid from blood plasma, since these also differ in ion composition (Wangemann 2006). The requirements of this boundary, and the implications of its failure, will be considered below.
3.5.2.2 How Constant Is the EP, and What Sets It?
If hearing thresholds depend on the EP, then should not the EP be highly constrained? How does the EP “know” what to be, and how constant is it for any normal individual? Regarding the former, no one has claimed to find a clear “reference” within the circuit that generates the EP. Regarding the latter, there are presently no data, since EP measurement is a terminal procedure conducted at a single point in time. We know that the EP varies among species (e.g., Conlee and Bennet 1993; Schmiedt 1993; Ohlemiller 2009) and also from cochlear base to apex, the EP being ~10 mV lower in the apex than in the base (Conlee and Bennet 1993; Ohlemiller et al. 2006, 2010). In guinea pigs, the spatial gradient correlates with endolymph Ca++ levels (Gill and Salt 1997), suggesting that Ca++ plays a role in EP regulation. For these things not to matter for thresholds, it might be necessary to offset EP reductions with compensatory increases in hair cell transducer conductance or in the gain of the cochlear amplifier. Modest reductions in the cochlear apex versus the base may have little effect, based on evidence that the cochlear amplifier plays a smaller role in setting thresholds at low frequencies than at high frequencies (Sewell 1984).
Patuzzi (Patuzzi 2011b) suggests that some type of communication must match the “current source” that is the stria with the “current load” that is the organ of Corti. Known regulatory points for K+ entry and exit from the scala media include the ATP-regulated KCNQ1/KCNE1 channel assemblies on the luminal surface of marginal cells and ATP-gated K+ channels that line the reticular lamina and spiral prominence (Mockett et al. 1994; Lee et al. 2001). The stria releases ATP plus probably other signal molecules as well. Activation of ATP-gated channels has been shown to reduce the EP and lower the input impedance of the scala media (Housley et al. 2013). The recent data of Housley et al. indicate that activation of these channels (with acute lowering of the EP) may be one component of noise-induced TTS and that genetic impairment of this process may promote PTS. The TTS contribution may also reflect ATP-mediated changes in the micromechanics of the organ of Corti (Bobbin 2001; Bobbin and Salt 2005). The widespread paracrine effects of ATP release from many cell types are diverse and complex. No single comprehensive picture has emerged of exactly purpose is served by ATP and ATP-triggered Ca++ waves in the lateral organ of Corti. What is presently not supported by any evidence is any signal that passes from the organ of Corti to the stria. Thus, any communication between the stria and organ of Corti appears to be one way.
In the sections that follow, we summarize what can be inferred about lateral wall injury, largely based on animal models. In assessing the contribution of the lateral wall to PTS, we are faced with two problems. First, what do we measure? The standard for injury to the organ of Corti (or its remediation) is hair cell density. We know that OHCs are closely linked to hearing thresholds. Anyone practiced in cochlear morphometry can tell in a single glance at an appropriately stained surface preparation of the organ of Corti whether there is hair cell loss. But the lateral wall is far more irregular, and any two normally hearing individuals may differ greatly with respect to fibrocyte density, capillary density, or strial thickness. In inbred animal models such as mice, differences in these metrics due to insults can be detected, yet the relation of such losses to hearing thresholds may not be easily discernible. Instead, the metric of choice is the EP, which constitutes a useful “one-stop-shopping” metric for assessing the status of the lateral wall. For the EP to be normal, a host of ions must be properly regulated, and a large set of boundaries must be maintained. At least in inbred mice, changes in the EP correlate well with lateral wall morphometry (Ohlemiller and Gagnon 2007; Ohlemiller et al. 2011). But this still leaves a second problem: Even if the EP is restored after injury (as it typically seems to be), this may not mean that all critical support functions of the lateral wall have been restored. Even temporary EP reduction may herald processes that promote permanent damage to the organ of Corti during the period that the EP was reduced. If this is the case, it may ultimately be difficult to separate lateral wall injury from organ of Corti injury as a primary cause of hearing loss or as a locus of its remediation. The chemistries of injury of the lateral wall and organ of Corti overlap extensively. Thus, finding the biochemical signature of oxidative or nitrosative stress in the lateral wall does not prove it is the critical site of injury. This is a frequent limitation of studies in this area (e.g., Hsu et al. 2000; Chen et al. 2008). Conditional knockout models for fibrocyte-specific proteins or of connexins restricted to the spiral ligament may help resolve these issues.
3.5.2.3 Noise and the Lateral Wall
While there is a moderate literature showing EP reduction by noise, few studies have dealt with EP recovery, and detailed parametric studies have been conducted only in mice (Hirose and Liberman 2003; Ohlemiller and Gagnon 2007; Ohlemiller et al. 2011). The most significant observation is that there is no one effect of a given exposure: Both acute and permanent EP changes depend on the exposure conditions, on the genetic background of the mice, and on their age. On some backgrounds (e.g., BALB/c), and in younger animals (<3 months), permanent EP reduction occurs more readily. Based on Fig. 3.4, the range of exposure levels that acutely reduce the EP may be strikingly broad, extending down to 92–95 dB SPL. Above 110–113 dB SPL, EPs appear uniformly low, typically below ~30 mV. In keeping with the observations of Hirose and Liberman (2003), this range may demarcate exposure levels that rupture the reticular lamina. This is a disruption the system almost certainly did not evolve to deal with, altering the shape of the organ of Corti and mixing endolymph and perilymph. When this occurs, loss of boundary integrity—not necessarily any injury or limitation of the lateral wall—will drive EP reduction. Moreover, the extent of PTS and pattern of cell loss will be determined by how the organ of Corti responds.
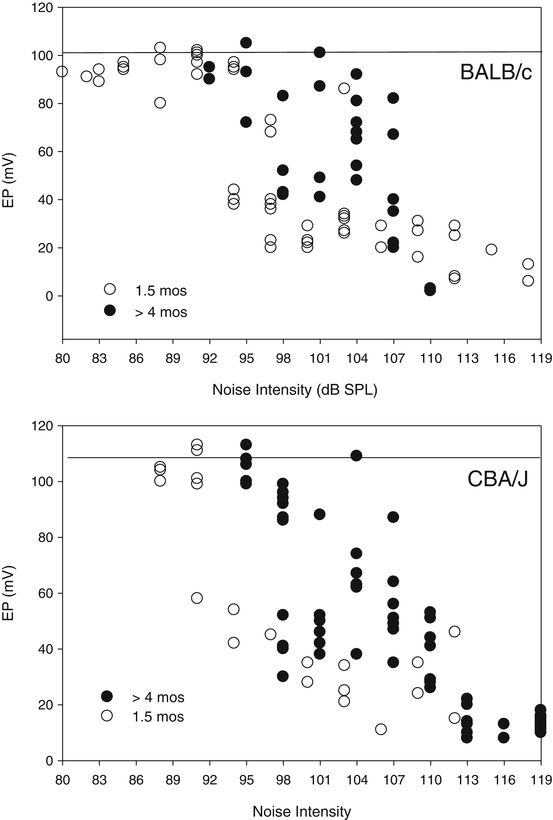
Fig. 3.4
Endocochlear potential is transiently reduced at surprisingly low noise levels. Basal turn endocochlear potential (EP) in young (1.5 months) or older (>4 months) BALB/c and CBA/J mice. Horizontal lines at tops of graphs indicate mean normal values for each strain. EPs were measured 1–3 h after 2 h of 8–16 kHz octave band noise at the intensities indicated. For each strain, the EP in younger mice was more vulnerable, but in each case, noise intensities causing acute EP reduction were surprisingly low (92–98 dB SPL). For both strains, exposures exceeding 110–113 dB SPL were associated with uniformly low EPs (<30 mV), likely indicating rupture of the reticular lamina
The data of Fig. 3.4 pose caveats for the design of experiments probing noise-induced PTS. Above and below the exposure intensity “rupture point,” both acute injury and its recovery may entail quite different processes. The critical question for the experimenter is “What do you want to know?” It is perhaps reasonable to assault the cochlea with 120 dB SPL noise if the experimental question is what can noise possibly do? But if the goal is to model, say, typical human occupational noise, then using supra-rupture noise may yield misleading results. It should be emphasized that, for any given noise type, the noise level required to rupture the reticular lamina varies by species (Fredelius et al. 1987; Fredelius 1988; Henderson et al. 1993, 2008; Hirose and Liberman 2003). Thus one cannot pick a “rupture point” noise level for mice that was established in, say, guinea pigs. Finally, the notion of noise as a cause of intermixing of endolymph and perilymph is not strictly limited to rupture. A lingering—yet still critical—debate pertains to the creation of “holes” in the reticular lamina that may be left by technically sub-rupture noise exposures when OHCs die. Such holes have been found in chinchillas (Bohne and Rabbit 1983), even for fairly low-level exposures (Harding and Bohne 2004), but so far not in other species. Although holes are eventually covered by scars formed by neighboring supporting cells, OHC death may be amplified by toxic levels of K+ while they persist.
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


